

 26
26
2025年值得关注的抗体药物:非癌症适应症
在这里,“值得关注的抗体药物”指的是到2025年底可能提交上市申请的抗体治疗药物。我们的预测基于所有证据,包括赞助公司或公司的公开披露,例如发布顶级临床研究数据以及正在进行的临床研究的预计主要完成日期。在我们识别的处于晚期临床研究阶段且用于非癌症适应症的抗体治疗药物中,我们预计到2025年底可能会提交13种药物的上市申请(表3)。以下按我们估计的上市申请提交日期顺序,给出了这些分子的关键信息。更多关于这些分子的数据可在补充表S1中找到。
Depemokimab(GSK)
Depemokimab(GSK3511294)是一种人源化单克隆IgG1κ抗体,靶向白细胞介素-5(IL-5),这是一种负责嗜酸性粒细胞的生长、激活和存活的细胞因子,也与2型炎症有关,通常通过血液嗜酸性粒细胞计数升高来检测。其Fc区域的M252Y、S254T、T256E(YTE)突变延长了半衰期,允许每年两次给药。由GSK开发的Depemokimab正在被研究用于治疗重度嗜酸性粒细哮喘。Depemokimab被授予用于治疗高嗜酸性粒细综合征的孤儿药认定。GSK计划在2024年底前在美国提交Depemokimab用于重度哮喘和伴有鼻息肉的慢性鼻窦炎(CRSwNP)的监管申请,随后将在欧盟、中国和日本提交监管申请。公司预计将在2025年推出双重适应症。
两项为期52周的随机、双盲、安慰剂对照的III期SWIFT-1(NCT04719832)和SWIFT-2(NCT04718103)研究评估了Depemokimab在有2型炎症的重度哮喘成人和青少年中的疗效和安全性。研究参与者(总计n = 762)按2:1的比例随机分配接受100 mg皮下注射Depemokimab或安慰剂,分别在第0周和第26周给药,此外还接受标准治疗(SOC)。主要终点是在52周内临床显著加重的年化率。在这两项试验中,Depemokimab使加重减少了54%。
2024年10月,GSK宣布了复制的随机、双盲III期ANCHOR-1(NCT05274750)和ANCHOR-2(NCT05281523)研究的积极顶线结果,这些研究评估了100 mg皮下注射Depemokimab在CRSwNP患者中的疗效和安全性。ANCHOR-1包括143名接受Depemokimab治疗的患者和128名接受安慰剂的患者,而在ANCHOR-2中,129名患者接受Depemokimab治疗,128名接受安慰剂。这两项试验均达到了其共同主要终点:1)在52周时从基线变化的总鼻内窥镜鼻息肉评分;2)从第49周到第52周的平均鼻塞评分从基线的变化。
GSK还在进行其他Depemokimab的III期研究,预计2025年将公布数据。之前参加过SWIFT试验之一的患者可以加入III期AGILE研究(NCT05243680),这是一项开放标签的12个月扩展研究,旨在描述Depemokimab作为重度未控制嗜酸性粒细哮喘成人和青少年受试者的辅助治疗的长期安全性、有效性和免疫原性特征。该研究的主要完成日期预计为2025年5月。
随机、双盲、非劣效性III期NIMBLE研究(NCT04718389)正在评估接受Depemokimab治疗的约1700名具有嗜酸性粒细表型的重度哮喘成人和青少年受试者的加重率、其他哮喘控制措施和安全性,与接受美泊利单抗或贝那利珠单抗治疗的患者相比。该研究的主要完成日期为2025年5月。此外,Depemokimab正在III期OCEAN研究(NCT05263934)中被研究用于160名接受标准治疗的复发性或难治性嗜酸性粒细肉芽肿性多血管炎(EGPA)成人患者,该研究的主要完成日期为2025年10月。
表3. 正在接受商业化赞助的、处于晚期临床研究阶段且用于非癌症适应症的单克隆抗体药物,预计在2024-2025年期间提交监管申请。
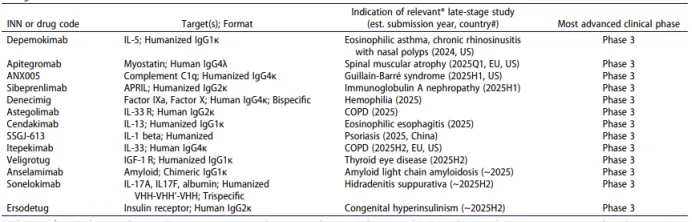
*预计提交监管申请的适应症。#首次提交上市申请的日期和国家是基于公司公告和关键试验完成日期的估计值。本表包含截至2024年12月1日公共领域中的信息。缩写:APRIL,增殖诱导配体;Fab,抗原结合片段;IL,白细胞介素;RSV,呼吸道合胞病毒。关于每种抗体的更多详细信息,请参阅补充表S1。关于处于晚期临床研究阶段的抗体治疗药物的更多数据,也可在以下网址查看:https://www.antibodysociety.org/antibodies-in-late-stage-clinical-studies/
Apitegromab(Scholar Rock)
Apitegromab(SRK-015)是由Scholar Rock开发的一种铰链稳定的IgG4λ单克隆抗体,用于抑制肌肉生长的负调节因子——肌肉生长抑制素(生长分化因子8)。该药物主要用于治疗脊髓性肌萎缩症(SMA),这是一种以运动神经元丢失和肌肉逐渐消耗为特征的疾病,同时也正在探索其在肥胖患者中保持瘦体肌肉质量的潜力。
美国食品药品监督管理局(FDA)已授予Apitegromab快速通道、孤儿药和罕见儿科疾病认定,欧洲药品管理局(EMA)也授予了优先药物(PRIME)和孤儿药产品认定,用于治疗SMA。Scholar Rock预计将在2025年第一季度在美国和欧盟提交上市申请。
最近报告了II期TOPAZ研究(NCT03921528)的结果,该研究评估了每四周一次(Q4W)静脉注射Apitegromab在晚发性2型和3型SMA患者中的安全性和有效性。共有58名参与者(平均年龄9.4岁),分为三个队列。第1队列(n = 23)包括能够行走的参与者,他们单独接受Apitegromab 20 mg/kg或与Nusinersen联合使用;第2队列(n = 20)包括不能行走的患者,接受与第1队列相同剂量的Apitegromab和Nusinersen;第3队列(n = 20)评估了两种剂量的Apitegromab(2 mg/kg和20 mg/kg)与Nusinersen联合使用。主要终点是通过扩展的Hammersmith功能性运动量表(HFMSE)的变化来衡量的运动功能改善。在第12个月时,第3队列观察到最显著的改善,2 mg/kg和20 mg/kg剂量的平均得分分别增加了5.3和7.1分。在研究的开放标签扩展期间,患者每四周一次静脉注射20 mg/kg的Apitegromab,12个月时观察到的益处在36个月时得以维持。
TOPAZ研究的结果为设计随机、双盲、安慰剂对照的5臂III期SAPPHIRE研究(NCT05156320)提供了依据,该研究共纳入了188名正在接受批准的存活运动神经元上调疗法(Nusinersen或Risdiplam)的晚发性SMA患者。2至12岁的2型和不能行走的3型SMA患者为主要疗效人群,他们接受每四周一次静脉注射Apitegromab 10 mg/kg、20 mg/kg或安慰剂,为期52周。探索性亚人群(筛选时年龄为13至21岁)的患者随机接受每四周一次的Apitegromab 20 mg/kg或安慰剂,最长可达52周。2024年10月宣布,该研究的主要终点——12个月时从基线变化的平均HFMSE——已达成。在主要疗效人群中,接受Apitegromab 10 mg/kg和20 mg/kg(n = 106)治疗的患者与接受安慰剂(n = 50)治疗的患者相比,从基线变化的HFMSE平均差异为1.8分(p = 0.0192)。接受20 mg/kg剂量Apitegromab治疗的患者(n = 53)与安慰剂相比显示出1.4分的平均差异(p = 0.1149)。在完成TOPAZ或SAPPHIRE研究的SMA患者中,正在进行一项开放标签的III期扩展研究(ONYX,NCT05626855),以评估每四周一次静脉注射20 mg/kg的Apitegromab的长期安全性和有效性,该研究为期104周。
ANX005(Annexon Biosciences, Inc.)
ANX005是由Annexon Biosciences开发的一种人源化抗C1q IgG4κ抗体。通过抑制C1q,ANX005旨在防止补体级联反应的激活,这可能导致炎症、细胞损伤和神经退行性病变。这种抑制旨在减轻如吉兰-巴雷综合征(GBS)等肌肉疾病中的补体介导的损伤。ANX005已获得FDA的快速通道和孤儿药认定,以及EMA的孤儿药产品认定,用于治疗GBS。Annexon计划在2025年上半年向FDA提交生物制品许可申请(BLA)。
Annexon报告了一项随机、安慰剂对照的III期试验(NCT04701164)的积极顶线结果,该试验评估了30 mg/kg或75 mg/kg静脉注射ANX005与安慰剂在241名GBS患者中的效果。该研究在吉兰-巴雷综合征高发且对标准治疗(静脉免疫球蛋白)接触有限的孟加拉国和菲律宾进行。结果显示,单次输注30 mg/kg的ANX005达到了研究的主要终点,在第8周时吉兰-巴雷综合征残疾量表(GBS-DS)改善了2.4倍。ANX005还导致神经丝轻链水平早期降低,这是一种神经损伤的生物标志物,在第2至第4周之间与安慰剂相比降低了11.2%。
根据FDA的反馈,Annexon与国际吉兰-巴雷综合征结果研究(IGOS,NCT01582763)启动了一项真实世界证据(RWE)协议,以比较III期研究参与者与西方患者的结果。公司预计将在2024年底前获得RWE可比性协议的初步顶线数据。
Sibeprenlimab(Otsuka Pharmaceutical Co., Ltd.)
Sibeprenlimab(VIS649)是一种人源化IgG2κ抗体,靶向增殖诱导配体(APRIL)抗原,该抗原与IgA肾病(IgAN)的发病机制有关。通过结合APRIL,该抗体阻断其受体BCMA和跨膜激活剂钙调蛋白和环孢素配体相互作用者的信号传导,从而干扰下游活动,如B细胞增殖、IgA产生和终末B细胞存活。FDA授予Sibeprenlimab突破性疗法认定,用于治疗IgAN。基于III期VISIONARY研究的积极结果,Otsuka计划在2025年上半年向FDA提交Sibeprenlimab用于治疗成人IgAN的生物制品许可申请。
随机、双盲、安慰剂对照的III期VISIONARY研究(NCT05248646)正在评估Sibeprenlimab(每四周一次皮下注射400 mg)与安慰剂在约530名接受标准治疗的IgAN患者中的效果。该研究的主要终点是治疗9个月后24小时尿液收集中的尿蛋白与肌酐比值(uPCR)从基线的相对变化。2024年10月宣布,该研究在预定的中期分析中达到了其主要终点。
Denecimig(诺和诺德)
Denecimig(NN-7769,NNC0365–3769,Mim8)是一种由Genmab的Duobody技术衍生的人源、铰链稳定的IgG4κ双特异性抗体,正在开发用于治疗血液凝固和凝血障碍。由诺和诺德开发,Denecimig促进活化的凝血因子IXa(FIXa)和X(FX)在血小板膜上的组装,模拟因子VIII,使其成为治疗A型血友病的潜在疗法,A型血友病是由编码因子VIII的F8基因突变引起的。Denecimig获得了FDA授予的孤儿药认定,用于治疗A型血友病。诺和诺德计划在2025年向监管机构提交Denecimig的上市申请。
诺和诺德的III期FRONTIER临床项目正在评估Denecimig作为预防性治疗A型血友病(无论是否存在抑制剂)的效果。该项目包括:
FRONTIER 2(NCT05053139):针对12岁及以上人群的研究。
FRONTIER 3(NCT05306418):针对儿童患者的研究。
FRONTIER 4(NCT05685238):开放标签扩展研究,旨在收集参与FRONTIER II期和III期研究后的长期安全数据。
FRONTIER 5(NCT05878938):研究从emicizumab转换为Denecimig对A型血友病患者的影响。
2024年5月,诺和诺德宣布,26周开放标签、随机的FRONTIER 2 III期研究达到了其共同主要终点,证明每周一次和每月一次的Denecimig剂量显著减少了12岁及以上A型血友病患者的治疗性出血事件数量。对于没有接受过预防性治疗的患者,每周Denecimig减少了97%的治疗性出血,每月剂量减少了99%。此外,每周治疗方案的86%患者和每月治疗方案的95%患者经历了零次治疗性出血,而没有预防性治疗的组中没有患者经历零次出血。在已经接受凝血因子预防性治疗的患者中,每周Denecimig剂量减少了48%的治疗性出血,每月剂量减少了43%。此外,每周治疗方案的66%患者和每月治疗方案的65%患者报告没有治疗性出血,显示出Denecimig优于先前预防性方法的优越性。
Astegolimab(Hoffmann-La Roche, Amgen)
Astegolimab(RO7187807,MSTT1041A,RG6149,AMG282)是一种人源单克隆IgG2κ抗体,靶向并抑制IL-33受体(也称为ST2)。IL-33是一种参与炎症反应的细胞因子,与多种免疫介导的疾病有关,如哮喘和慢性阻塞性肺病(COPD)。2016年,Amgen将Astegolimab的全球独家权利授权给Genentech,Genentech是Roche的独立子公司,计划在2025年提交COPD的监管申请。
Roche正在开展两项Astegolimab的III期研究,针对COPD患者。随机、双盲、安慰剂对照的ARNASA研究(NCT05595642)正在评估Astegolimab在1290名COPD患者中的疗效和安全性。在这项三臂研究中,参与者接受每两周一次(Q2W)或每四周一次(Q4W)的皮下注射Astegolimab,或每两周一次的安慰剂。主要研究终点是52周治疗期间中度和重度COPD加重的年化率。预计主要完成日期为2025年6月。
III期开放标签扩展ALNASA研究(NCT05878769)旨在评估完成III期ARNASA或IIb期ALIENTO(NCT05037929)研究中52周安慰剂对照治疗期的COPD患者中Astegolimab的长期安全性和进一步探索其疗效。预计2000名患者将接受每两周一次的皮下注射Astegolimab,直至研究结束。主要结果测量是研究治疗最后一次给药后12周内所有不良事件的发生率。预计主要完成日期为2027年6月。
Cendakimab(Bristol Myers Squibb)
Cendakimab(CC-93538,RPC4046;ABT-308,13C5.5,BMS986355)是一种人源化IgG1κ抗体,阻断IL-13与其两个受体IL-13 Rα1和IL-13 Rα2的结合。抗体的Fc区域经过工程改造,以降低Fc介导的效应功能(L234A,L235A)。Cendakimab由多家公司开发,用于免疫介导的疾病。2013年,AbbVie将Cendakimab授权给Receptors,后者于2015年被Celgene收购。Celgene于2019年被Bristol Myers Squibb(BMS)收购。一项III期试验评估了Cendakimab在嗜酸性粒细食管炎(EoE)患者中的疗效和安全性,达到了两个共同主要终点,为2025年的上市申请提供了可能。
在一项随机、双盲、安慰剂对照的III期研究(NCT04753697)中,评估了Cendakimab在成人和青少年EoE患者中的疗效和安全性。该研究有三个臂:1)Cendakimab 360 mg每周一次皮下注射,持续24周,随后Cendakimab 360 mg每周一次皮下注射,再持续24周;2)Cendakimab 360 mg每周一次皮下注射,持续24周,随后Cendakimab 360 mg每两周一次皮下注射,持续24周,在维持阶段的交替周给予匹配的安慰剂以保持盲态;3)匹配的安慰剂每周一次皮下注射,持续24周,随后匹配的安慰剂每周一次皮下注射,再持续24周。该研究达到了共同主要终点,即在24周时吞咽困难日临床反应和嗜酸性粒细组织学反应的变化,与安慰剂相比,在症状(吞咽困难日)和食管嗜酸性粒细计数方面显示出统计学显著的减少。
SSGJ-613(Sunshine Guojian Pharmaceutical (Shanghai) Co. Ltd., 3SBio)
SSGJ-613是一种人源化单克隆抗体,靶向IL-1β,这是急性痛风发作的关键介质。该分子由Sunshine Guojian Pharmaceutical (Shanghai) Co. Ltd.和3SBio开发,目前正在进行治疗痛风和痛风性关节炎的临床研究。3SBio计划在2025年底前向NMPA提交治疗急性痛风性关节炎的新药上市申请。
最近报告了一项在中国急性痛风性关节炎患者中进行的活性对照II期研究的积极结果。在这项研究中,共纳入了90名受试者,符合条件的患者按1:1:1的比例随机接受单剂量(200或300 mg皮下注射)的SSGJ-613或甲泼尼龙(1 mL肌肉注射)。研究表明,无论哪种剂量,单次注射SSGJ-613都能实现与类固醇相似的快速止痛效果,并能有效预防发作。
2024年1月,Sunshine Guojian Pharmaceutical启动了一项活性对照III期研究(CTR20233982,NCT06169891,SSGJ-613-AG-III-01),评估SSGJ-613抗体注射在中国急性痛风患者中的疗效和安全性。随机患者在第1天接受一次SSGJ-613(200 mg)皮下注射和匹配的安慰剂(0.9%氯化钠),或匹配SSGJ-613的甲泼尼龙(肌肉注射)。预计招募500名受试者。主要结果测量包括从基线到给药后72小时目标关节疼痛强度的变化(使用0–100 mm视觉模拟量表(VAS)测量)以及首次新发作的时间(在首次给药后12周内测量)。
Itepekimab(Sanofi, Regeneron)
Itepekimab(SAR440340,REGN3500)是一种利用Regeneron专有的VelocImmune技术开发的人IgG4κ单克隆抗体。该抗体结合并抑制IL-33的信号传导,IL-33是气道炎症的启动因子和放大因子。Itepekimab由Regeneron和Sanofi联合开发,正在研究用于治疗呼吸系统疾病。Itepekimab的主要适应症是慢性阻塞性肺病(COPD),Sanofi计划在2025年下半年在美国和欧盟提交上市申请。
Itepekimab正在两项随机、双盲、安慰剂对照的III期研究AERIFY-1和AERIFY-2(NCT04701983和NCT04751487)中进行研究。三臂AERIFY-1研究旨在评估皮下注射Itepekimab与安慰剂在中重度慢性阻塞性肺病的前吸烟者中的疗效和安全性。受试者按1:1:1的比例随机接受Itepekimab 300 mg每两周一次(Q2W)、Itepekimab 300 mg每四周一次(Q4W)与每两周一次的匹配安慰剂交替给药,或每两周一次的匹配安慰剂,最长可达52周。AERIFY-2研究设计相同,但纳入了当前吸烟者和前吸烟者,均为中重度COPD患者。AERIFY-1和AERIFY-2的预计招募人数分别为960人和1210人。两项研究的主要终点均为COPD中度或重度急性加重的年化率(AECOPD),在安慰剂对照治疗期间测量。AERIFY-1和AERIFY-2的主要完成日期预计分别为2025年6月27日和5月30日。
Itepekimab的长期安全性和耐受性正在双盲、两臂III期AERIFY-4研究(NCT06208306)中进行评估。这项扩展研究将从参与AERIFY-1和AERIFY-2研究的受试者中招募约700名受试者,旨在生成额外的安全性数据,评估治疗反应的持久性,并提供Itepekimab皮下注射每两周一次(Q2W)或每四周一次(Q4W)的额外药代动力学(PK)和免疫原性评估。AERIFY-4的主要完成日期预计为2026年12月。
Veligrotug(Viridian Therapeutics, Inc.)
Veligrotug(VRDN-001,AVE-1642)是一种人源化IgG1κ单克隆抗体,靶向胰岛素样生长因子1受体(IGF1R)的细胞外域。最初由ImmunoGen与Sanofi-Aventis合作开发,Viridian从ImmunoGen获得了除放射性药物外所有非肿瘤适应症的全球独家开发和商业化权利。Viridian预计将在2025年下半年提交Veligrotug用于治疗甲状腺眼病(TED)的生物制品许可申请(BLA)。
2024年9月,Viridian Therapeutics宣布了安慰剂对照III期THRIVE研究(NCT05176639)的积极顶线结果,该研究首次在健康志愿者和中重度活动性TED患者中评估了Veligrotug的多剂量递增(MAD),随后在TED患者中评估了Veligrotug的单剂量水平。在研究的MAD部分,受试者被随机分配接受30分钟静脉输注Veligrotug,剂量范围为3 mg/kg至20 mg/kg(n = 75)或安慰剂(n = 38)。TED患者随后被随机分配接受每3周一次的10 mg/kg Veligrotug静脉输注或安慰剂。研究的主要终点是MAD TED受试者在第6周和III期研究受试者在第15周的突眼反应率。在给予Veligrotug五次输注后的15周,研究达到了主要和所有次要终点,显示出在所有测量的TED体征和症状上具有高度统计学显著性(p < 0.0001)的改善。在接受Veligrotug治疗的患者中,突眼反应率为70%,67%实现了总体反应,而接受安慰剂的患者中,突眼反应率为5%,5%实现了总体反应。
Veligrotug的随机、双盲、安慰剂对照III期研究THRIVE-2(NCT06021054)在慢性TED患者中已超额完成招募。在这项研究中,患者将接受5次10 mg/kg Veligrotug静脉输注。预计将在2024年底前公布顶线数据。
Anselamimab(AstraZeneca)
Anselamimab(CAEL-101,11-1F4)是一种嵌合单克隆IgG1κ抗体,旨在靶向由骨髓中的浆细胞产生的错误折叠的轻链蛋白形成的淀粉样纤维,并通过巨噬细胞和中性粒细胞介导吞噬作用。这些纤维在组织和器官中积聚,导致一种称为轻链(AL)淀粉样变性的疾病。这种罕见疾病导致淀粉样物质在心脏、肾脏和肝脏等关键器官中沉积,逐渐损害其功能,并随时间造成显著的器官损伤。
Fortress Biotech, Inc.的子公司Caelum Biosciences从哥伦比亚大学获得了Anselamimab的全球独家许可,并随后与Alexion Pharmaceuticals, Inc.合作开发该分子。AstraZeneca在2021年收购了Alexion和Caelum Biosciences。Anselamimab获得了FDA和EMA分别授予的孤儿药和孤儿药产品认定,作为AL淀粉样变性患者的潜在疗法。FDA还授予了Anselamimab用于AL淀粉样变性的快速通道认定。AstraZeneca预计将在2025年公布Anselamimab在AL淀粉样变性的关键III期研究结果,这可能允许在2025年提交Anselamimab用于AL淀粉样变性的监管申请。
AstraZeneca的Alexion部门目前正在Cardiac Amyloid Reaching for Extended Survival(CARES)项目中评估Anselamimab,该项目旨在通过两项III期研究评估其在AL淀粉样变性患者中的疗效与安慰剂相比。两项研究的主要终点均为从试验药物首次给药至死亡或试验结束的时间。随机、盲法III期NCT04512235研究评估了281名未经治疗的Mayo IIIa期AL淀粉样变性患者中静脉注射Anselamimab或安慰剂联合标准治疗(SOC)的疗效和安全性。该研究的主要完成日期预计为2025年4月。III期NCT04504825研究设计相同,但包括125名Mayo IIIb期患者。该研究的主要完成日期预计为2025年5月。
Sonelokimab(Merck Serono, MoonLake Immunotherapeutics AG)
Sonelokimab(M1095,ALX-0761)是一种三特异性抗体片段,靶向IL-17A、IL-17F和白蛋白。该分子由三个纳米抗体组成,这些纳米抗体是骆驼科来源的仅重链单域抗体,通过柔性的甘氨酸-丝氨酸间隔物共价连接。特异性靶向IL-17F的结构域和特异性靶向IL-17A及IL-17F的结构域之间,通过靶向白蛋白的结构域分隔,以促进纳米抗体在炎症水肿部位的富集。2021年,MoonLake Immunotherapeutics AG从Merck KGaA获得了Sonelokimab的许可,Merck KGaA于2013年通过与Ablynx(现为Sanofi公司旗下的Ablynx)的全球开发和商业化协议获得了Sonelokimab的全部独家权利。
MoonLake Immunotherapeutics正在开发Sonelokimab用于治疗化脓性汗腺炎(HS)和银屑病关节炎(PSA),以及皮肤科和风湿病学领域的其他适应症。在2023年宣布的全球II期MIRA试验的积极顶线结果之后,公司启动了III期VELA项目,预计将招募800名中重度HS患者。主要终点的读出预计在2025年中期;积极的研究结果可能使公司能够在2025年底之前提交上市申请。
VELA项目包括两项III期试验,VELA-1(NCT06411899)和VELA-2(NCT06411379),这些试验是全球性、随机、双盲、安慰剂对照的研究,设计相同,评估皮下注射Sonelokimab与安慰剂在中重度HS成年受试者中的疗效和安全性。随机分配到Sonelokimab组的患者将在第0至6周接受每两周一次(Q2W)的120 mg Sonelokimab,然后从第8周开始至第48周每四周一次(Q4W)。安慰剂组的患者将在第0至6周接受每两周一次的安慰剂,然后从第8周开始至第16周每四周一次,之后他们将接受120 mg Sonelokimab每两周一次,共4剂,从第16周至第22周,然后从第24周至第48周每四周一次。主要终点是更高的临床反应测量值,即HS临床反应(HiSCR)75,定义为从基线减少至少75%的脓肿和炎症结节(AN)计数,且脓肿或引流瘘管计数无增加,该指标在第16周测量。每项研究预计招募400名患者,主要完成日期预计在2025年6月。
Sonelokimab也在银屑病关节炎(PSA)中进行研究,II期ARGO试验已达到主要和关键次要终点,预计在2024年第四季度启动III期试验(NCT06641076,NCT06641089)。预计在2024年底前启动掌跖脓疱病和放射学及非放射学轴性脊柱关节炎(axSpA)的II期试验。
Ersodetug(Rezolute, Inc.)
Ersodetug(RZ358,XOMA 358)是一种人源IgG2κ单克隆抗体,靶向胰岛素受体。该抗体结合胰岛素受体上的一个变构位点,在目标组织(如肝脏、脂肪和肌肉)中调节胰岛素的结合、信号传导和活性,以将血糖水平恢复到正常范围。在小鼠和健康人类中,Ersodetug已被证明可诱导全身性胰岛素抵抗,并逆转胰岛素刺激的低血糖,其有潜力用于治疗任何形式的高胰岛素血症(HI)引起的低血糖,HI是一种罕见的、遗传性的、儿科内分泌疾病,具有先天性和获得性形式。
Rezolute, Inc.于2017年从XOMA Corporation获得了Ersodetug的全球开发和商业化权利,正在开发该分子用于治疗高胰岛素血症(HI)。Ersodetug已获得美国和欧盟的孤儿药认定,以及美国的儿科罕见病认定。Ersodetug在先天性HI患者中的III期sunRIZE研究的主要完成日期预计在2025年4月,顶线数据预计在2025年下半年公布。
随机、双盲、安慰剂对照、平行臂III期sunRIZE试验(NCT06208215)正在评估Ersodetug在先天性HI患者中的疗效和安全性,作为标准治疗(SOC)的附加疗法,与单独的SOC疗法相比,为期24周,以及在随后的开放标签扩展期间Ersodetug的长期安全性和疗效。该研究预计将招募48名受试者(年龄≥1岁至≤45岁),按1:1的比例随机分配到两个剂量组(5或10 mg/kg)。每个剂量水平内的受试者将进一步按2:1的比例随机分配,接受Ersodetug作为SOC的附加疗法或安慰剂作为SOC的附加疗法。还将为年龄≥3个月至<1岁的受试者开展一个平行的开放标签组,招募8名受试者。在关键治疗期(24周)完成后,患者可能会转入开放标签扩展期。主要终点是通过即时护理自我监测血糖从基线变化的平均每周低血糖事件数。
Erfonrilimab(江苏康宁杰瑞生物制药有限公司)Erfonrilimab(KN046)是一种具有2 + 2对称设计((VHH-VHH')2-Fc)的人源化双特异性抗体,靶向免疫系统检查点蛋白PD-L1和CTLA-4。江苏康宁杰瑞正在开发Erfonrilimab用于多种实体瘤,并与多家公司就Erfonrilimab开展了合作,包括辉瑞、泽璟、东阳光、开拓、信诺维和先声药业。Erfonrilimab获得了美国食品药品监督管理局(FDA)授予的用于胸腺上皮肿瘤和胆管癌的孤儿药资格。江苏康宁杰瑞预计将在2024年完成对鳞状(sq)非小细胞肺癌(NSCLC)一线治疗的最终总生存期(OS)分析,如果结果积极,可能会向中国国家药品监督管理局(NMPA)提交新药上市申请(NDA)。
近期报告了Erfonrilimab联合化疗作为转移性非小细胞肺癌(NSCLC)一线治疗的开放标签2期KN046–202试验(NCT04054531)的结果。该研究包括两个队列:1)非鳞状NSCLC患者接受培美曲塞联合KN046(5 mg/kg静脉注射,每3周一次)和卡铂(n = 51);2)鳞状NSCLC患者接受紫杉醇联合KN046(5 mg/kg静脉注射,每3周一次)和卡铂(n = 36)。经过四个周期后,非鳞状NSCLC的维持治疗为KN046联合培美曲塞,鳞状NSCLC的维持治疗为KN046。主要终点为确认的客观缓解率(ORR)和中位缓解持续时间(DoR)。非鳞状NSCLC和鳞状NSCLC队列的确认ORR分别为43.1%和50.0%。非鳞状NSCLC和鳞状NSCLC队列的中位DoR分别为9.7个月(95% CI:4.01–20.73)和7.3个月(95% CI:3.52-未达到)。非鳞状NSCLC和鳞状NSCLC队列的中位无进展生存期(PFS)分别为5.8个月(95% CI:4.80–7.16)和5.7个月(95% CI:4.17–8.71)。非鳞状NSCLC队列的中位总生存期(OS)为27.2个月(95% CI:15.18-未达到),鳞状NSCLC队列的中位OS为26.6个月(95% CI:12.19-未达到)。
正在进行的随机、安慰剂对照的3期ENREACH-LUNG-01研究(NCT04474119,CTR20201294)旨在验证KN046联合含铂化疗在晚期鳞状NSCLC患者一线治疗中的疗效和安全性。在这项2臂研究中,患者在双盲期的前12周接受卡铂、紫杉醇和静脉注射KN046 5 mg/kg每3周一次,然后在交叉期接受静脉注射KN046 5 mg/kg每2周一次;或者患者接受卡铂、紫杉醇和安慰剂。该研究于2020年9月启动,预计招募482名患者。主要终点是根据实体瘤疗效评价标准1.1版(RECIST 1.1)的无进展生存期(PFS),次要终点是在评估期间(3年)的总生存期(OS)。
表4. 预计在2024-2025年进行监管申报的、由商业资助的处于晚期临床研究阶段的抗癌单克隆抗体。
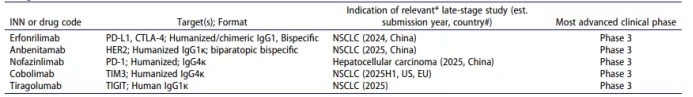
*预计进行监管申报的适应症。#首次上市申请提交日期和国家是根据公司公告及关键试验完成日期估算的。本表包含的信息截至2024年12月1日均为公开领域信息。缩写:ADC,抗体-药物偶联物;BCMA,B细胞成熟抗原;CEACAM5,癌胚抗原相关细胞黏附分子-5;CTLA-4,细胞毒性T淋巴细胞抗原-4;HER,人表皮生长因子受体;PD-L1,程序性细胞死亡蛋白配体1;TIGIT,T细胞免疫受体含Ig和ITIM结构域;TIM-3,T细胞免疫球蛋白和黏蛋白结构域含有蛋白-3。更多关于每种抗体的详细信息请参阅补充表S1。处于晚期临床研究阶段的其他研究性抗体治疗药物的额外数据也可用。
Anbenitamab(上海津曼特生物科技有限公司,江苏康宁杰瑞生物制药有限公司)
安本尼单抗(KN026)是一种人源化双表位双特异性IgG1κ单克隆抗体,靶向HER2的两个非重叠表位。该抗体采用通用轻链设计,重链通过“突起-孔洞”和静电导向突变进行工程改造,以实现异二聚体化。通过阻断HER2,安本尼单抗能够抑制肿瘤细胞生长,并促进抗体依赖性细胞介导的细胞毒性(ADCC)。
2021年,上海津曼特从江苏康宁杰瑞获得了安本尼单抗在中国大陆用于乳腺癌和胃癌适应症的独家开发和商业化许可权,包括作为单药以及与KN046(一种靶向免疫检查点蛋白PD-L1和CTLA-4的双特异性抗体)联合用药。安本尼单抗联合化疗被中国国家药品监督管理局(NMPA)授予突破性疗法认定,用于治疗对一线标准治疗无效的HER2阳性胃癌患者。此外,美国食品药品监督管理局(FDA)授予安本尼单抗联合KN046用于治疗HER2阳性或低表达的胃/胃食管结合部(G/GEJ)癌的孤儿药资格。预计将在2025年提交安本尼单抗的新药上市申请(NDA)。
上海津曼特正在开展关键性试验,评估安本尼单抗在胃癌和乳腺癌中的疗效。KN026–001(NCT05427383,CTR20213458)是一项两阶段随机、2/3期临床研究,旨在评估静脉注射安本尼单抗(30 mg/kg,每3周一次)联合化疗(紫杉醇/多西他赛/伊立替康)与安慰剂联合化疗相比,在约286例HER2阳性晚期不可切除或转移性胃癌患者(包括胃食管结合部腺癌)中的疗效,这些患者对一线治疗无效。该研究的主要终点是无进展生存期(PFS)和总生存期(OS),最长随访时间为2.5年。预计该研究的主要完成日期为2025年11月。
上海津曼特还在开展一项随机、对照、开放标签的3期KN026–001试验(NCT05838066,CTR20231442),评估安本尼单抗联合HB1801(白蛋白结合型多西他赛)与曲妥珠单抗+帕妥珠单抗+多西他赛相比,在一线治疗HER2阳性复发或转移性乳腺癌患者中的疗效和安全性。该研究的主要终点是PFS,预计主要完成日期为2026年7月。
Nofazinlimab(基石药业,三生国健药业股份有限公司)
诺法津单抗(CS1003)是一种人源化、铰链区稳定的IgG4κ单克隆抗体,靶向人类PD-1,由基石药业的专有杂交瘤平台开发。诺法津单抗可与人和小鼠PD-1发生交叉反应,便于在同种小鼠肿瘤模型中进行疗效测试。目前,该药物由基石药业和三生国健共同开发用于多种肿瘤的免疫治疗,其中三生国健拥有在中国大陆开发、注册、生产和商业化诺法津单抗的权利,而基石药业保留中国大陆以外的权利。诺法津单抗被美国授予用于治疗肝细胞癌(HCC)患者的孤儿药资格。三生国健计划于2025年向中国国家药品监督管理局(NMPA)提交诺法津单抗用于HCC的新药上市申请(NDA)。
双盲、随机的3期CS1003–305研究(NCT04194775,CTR20192524)正在评估静脉注射诺法津单抗联合仑伐替尼(一种标准治疗的酪氨酸激酶抑制剂)与安慰剂联合仑伐替尼相比,在晚期HCC患者一线治疗中的疗效和安全性。参与者接受200 mg诺法津单抗每3周一次联合仑伐替尼,或安慰剂联合仑伐替尼,直至疾病进展或出现不可耐受的毒性,或因其他原因退出研究。该研究的主要终点是总生存期(OS)。计划招募534名患者,预计最终OS分析将于2025年上半年完成。
Cobolimab(GSK)
科博利单抗(GSK4069889,TSR-022)是一种人源化、铰链区稳定的IgG4κ单克隆抗体,靶向免疫检查点T细胞免疫球蛋白和黏蛋白结构域蛋白-3(TIM-3),TIM-3与抑制抗肿瘤反应相关。通过抑制TIM-3,科博利单抗激活免疫系统功能,且与PD-1药物联合使用时活性增强。科博利单抗由TESARO发现并初步开发,GSK于2019年收购了TESARO。GSK计划于2025年上半年在美国和欧盟提交科博利单抗用于二线非小细胞肺癌(NSCLC)患者的监管申请。
随机、开放标签、三臂的2/3期COSTAR Lung试验(NCT04655976)旨在比较1)科博利单抗联合抗PD-1药物多塔利单抗+多西他赛,2)多塔利单抗+多西他赛,3)单独多西他赛的效果。该研究已招募758名先前接受过抗PD-(L)1治疗和化疗后进展的晚期NSCLC患者,分别随机分配到3个研究组(比例为2:1:1)。该研究的主要终点是接受联合治疗与单独多西他赛治疗的参与者总生存期(OS)的比较,最长随访时间为44个月。预计数据将在2025年上半年公布。
Tiragolumab(Hoffmann La Roche)Tiragolumab(RO7092284,RG6058,MTIG7192A)是一种靶向免疫检查点蛋白T细胞免疫受体免疫球蛋白和ITIM结构域(TIGIT)的人IgG1κ抗体。TIGIT存在于激活的T细胞、自然杀伤细胞和调节性T细胞上,是一种免疫系统功能的抑制因子。在一些临床前和临床研究中,TIGIT和PD-1/PD-L1的双重阻断显示出增强的抗癌效果。罗氏正在评估tiragolumab与抗PD-L1药物阿替利珠单抗(Tecentriq®)的联合治疗方案,用于治疗肺癌、食管癌和肝细胞癌(HCC)的多项晚期研究。截至2024年第三季度,罗氏预计可能在2025年提交tiragolumab和阿替利珠单抗联合治疗方案的上市申请,用于治疗接受至少两个周期的同步铂类化疗放疗且未出现放射学疾病进展的III期不可切除非小细胞肺癌(NSCLC)患者,该申请基于SKYSCRAPER-03研究(NCT04513925);以及用于治疗局部晚期不可切除或转移性PD-L1选择的非小细胞肺癌(NSCLC)患者,这些患者没有表皮生长因子受体(EGFR)突变或间变性淋巴瘤激酶易位,该申请基于安慰剂对照的III期SKYSCRAPER-01研究(NCT04294810)。然而,SKYSCRAPER-01研究未能达到其主要终点无进展生存期(PFS)或总生存期(OS)。2024年7月,罗氏集团成员基因泰克报告称,安慰剂对照的II/III期SKYSCRAPER-06研究(NCT04619797)未能在主要分析中达到PFS的主要终点,以及在首次中期分析中未能达到OS的主要终点,该研究评估了tiragolumab与阿替利珠单抗和化疗联合用于非鳞状非小细胞肺癌的一线治疗。公司表示,他们打算终止该研究。
正在进行的其他晚期研究正在评估tiragolumab与其他疗法联合用于食管癌(SKYSCRAPER-07(NCT04543617)、SKYSCRAPER-08(NCT04540211))和肝细胞癌患者(SKYSCRAPER-14,NCT05904886)的一线治疗。在中国进行的安慰剂对照的III期SKYSCRAPER-08研究正在评估tiragolumab与阿替利珠单抗和化疗联合用于不可切除局部晚期、不可切除复发性或转移性食管癌患者的一线治疗。符合条件的患者以1:1的比例随机分配到两个研究组之一,分别接受:1)tiragolumab 600 mg + 阿替利珠单抗1200 mg + 化疗,或2)安慰剂 + 化疗。该研究达到了独立审查机构评估的PFS和OS两个主要终点,其中包含tiragolumab治疗组的中位PFS为6.2个月,安慰剂组为5.4个月;中位OS分别为15.7个月和11.1个月。
总体而言,2024年对于抗体治疗药物来说是非常好的一年,这体现在处于晚期临床开发阶段的分子数量显著增加(本文报告的有178个),与2010-2024年《值得关注的抗体》中报告的数量(范围为26-138)相比。此外,与2024年版《值得关注的抗体》相比,我们报告的处于监管审查阶段以及获得首次批准的抗体治疗药物的数量也略有增加。
然而,在这些积极消息的同时,我们也注意到一个令人担忧的问题,即首次提交给美国食品药品监督管理局(FDA)的生物制品许可申请(BLA)中,未获批准的比例有所增加,这延迟了患者获得治疗的机会。在美国监管审查中的9种研究性抗体(即那些在其他国家尚未获批的抗体)中,目前有4种处于第二次审查周期,此前它们收到了FDA发出的完整回复函(CRL)(见表2)。此外,FDA还在审查另外4种抗体治疗药物的申请,这些药物已在其他国家获得首次批准,因此未包含在表2中,其中3种收到了CRL。因此,目前处于FDA审查中的分子总数为13个,其中7个(54%)处于第二次审查周期(补充表S2)。我们最近注意到FDA发出的CRL数量有所增加,尤其是由于化学、生产和控制(CMC)问题导致的CRL(见图3)。当FDA对某一营销申请存在担忧时,会发出CRL。这些担忧必须由申请公司通过重新提交的BLA来解决,并且该BLA必须再次经过FDA的审查,这可能会使产品的批准延迟6个月或更久。值得注意的是,大多数CRL是由于CMC缺陷而发出的,而不是由于安全或有效性方面的担忧,这凸显了申办方及其第三方制造合作伙伴需要积极主动,确保制造场所符合FDA对现行良好生产规范的要求。
与以往一样,我们将继续跟踪抗体治疗药物进入晚期临床研究并逐步走向市场批准的进展。我们期待在未来《值得关注的抗体》文章中进一步报告相关趋势,并记录这些分子的成功。
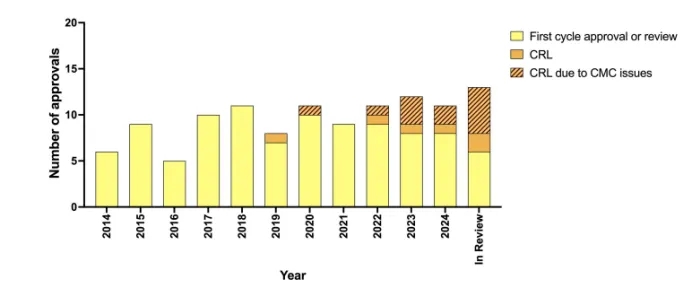
图3. 美国食品药品监督管理局发出完整回复函的趋势。黄色部分表示经过首次审查后获批的抗体治疗药物或目前正在进行首次审查的药物。橙色部分表示FDA发出的完整回复函(CRL)。带有条纹的橙色部分表示因化学、生产和控制(CMC)问题而由FDA发出的完整回复函。2024年的数据截至2024年12月7日。
更多阅读



长按屏幕识别二维码
打开手机扫描二维码




