2036
2036
2024年仅在中国或俄罗斯首次获批上市的药物
Mazorelvimab、Zamerovimab(Synermore Biologics Co. Ltd.)
Mazorelvimab和zamerovimab(克瑞毕®,SYN-023)是一种由两种人源化IgG1抗体CTB-011和CTB-012按1:1比例混合而成的药物,这两种抗体能够结合狂犬病外膜糖蛋白的两个非重叠表位。2024年6月,中国国家药品监督管理局(NMPA)批准了mazorelvimab和zamerovimab混合物用于狂犬病的暴露后预防治疗。
在一项随机III期研究(NCT04644484)中,mazorelvimab和zamerovimab混合物与人狂犬病免疫球蛋白(HRIG)被比较用于成人3级狂犬病暴露风险的暴露后预防。该研究在中国开展,共纳入了1000名受试者,他们接受了中国批准的狂犬病疫苗,并联合使用了干预措施,即研究性抗体混合物或对照药物HRIG。干预措施通过直接注入伤口给药,若无法直接注入伤口,则通过皮下(SC)或肌肉(IM)注射给药。
另一项II期临床研究(NCT03961555)的结果也已近期公布,该研究评估了mazorelvimab和zamerovimab混合物的效果,其研究设计与NCT04644484研究相似。NCT03961555研究共纳入448名受试者,他们被随机分配在可能暴露于狂犬病后的54小时内接受0.3 mg/kg SYN023或0.133 mL/kg HRIG以及狂犬病疫苗。结果显示,mazorelvimab和zamerovimab混合物与狂犬病疫苗(RabAvert/Rabipur)联合使用的抗狂犬病免疫反应不劣于HRIG与疫苗联合使用。在任何患者中均未发生可能/确诊的狂犬病病例,且mazorelvimab和zamerovimab混合物的安全性可接受。
Vunakizumab(江苏恒瑞医药股份有限公司)
Vunakizumab(SHR-1314,夫那奇珠单抗,安达静®)是一种人源化IgG1抗体,能够结合白细胞介素17A(IL-17A),这是一种在多种自身免疫性疾病相关的炎症过程中发挥关键作用的细胞因子。2024年8月20日,Vunakizumab获得NMPA批准,用于治疗适合接受系统治疗或光疗的中重度斑块状银屑病成人患者。
Vunakizumab的批准基于一项多中心、随机、双盲、平行、安慰剂对照的III期临床研究(SHR-1314-301)。共纳入690名中重度斑块状银屑病成人患者,并以2:1的比例随机分配到vunakizumab组(240 mg每两周一次,随后每四周一次)或安慰剂组(在第12周转换为vunakizumab 240 mg治疗,第12、14和16周每两周给药一次,之后每四周一次)。共同主要终点是第12周达到银屑病面积和严重程度指数90(PASI 90)的受试者比例以及第12周达到静态医生整体评估(sPGA)评分0/1的受试者比例。关键次要终点包括第12周达到PASI 75/PASI 100的受试者比例以及sPGA评分为0的受试者比例。在第12周,vunakizumab组的PASI 90反应率为76.8%,而安慰剂组为0.9%。同样,sPGA 0/1反应率为71.8%,而安慰剂组为0.4%。其他关键指标也显示出显著差异:PASI 75为93.6%对4.0%,PASI 100为36.6%对0.0%,sPGA 0反应率为38.2%对0.0%。在vunakizumab组中,疗效在第52周得以维持,PASI 100和sPGA 0的反应率分别达到63.1%和63.3%。Vunakizumab的临床反应起效迅速,PASI平均百分比在第2周就已降低超过50%。到第4周,56.6%的受试者已达到PASI 75反应。在12周诱导期内,接受vunakizumab治疗的受试者与接受安慰剂的受试者在不良事件的发生率和严重程度方面相似。
Xeligekimab(重庆智翔金泰生物制药股份有限公司)
Xeligekimab(GR1501,赛立奇单抗,金立希®)是一种人源化、铰链稳定的IgG4抗体,靶向白细胞介素-17A(IL-17A),这是一种参与多种免疫介导炎症疾病的细胞因子。2024年8月27日,智翔生物宣布Xeligekimab获得国家药品监督管理局(NMPA)批准,用于治疗适合接受系统治疗或光疗的中重度斑块状银屑病成人患者。2024年1月,NMPA接受了Xeligekimab用于放射学轴性脊柱关节炎适应症的新药上市申请(NDA)。
Xeligekimab的批准基于一项随机、双盲、安慰剂对照、多中心的III期临床研究(CTR20210246/ChiCTR2100043223),研究对象为中重度斑块状银屑病患者。患者随机分配到接受200 mg Xeligekimab皮下注射(Q2W,每两周一次,n = 281)或安慰剂(n = 139)组,持续12周,随后将治疗方案延长至Xeligekimab每四周一次(Q4W),再持续40周。到第12周时,Xeligekimab组中有90.7%的受试者从基线改善了至少75%,而安慰剂组为8.6%。此外,治疗组中有74.4%的患者在第12周时达到皮肤清晰或几乎清晰(PGA 0/1),而安慰剂组为3.6%。主要终点成功达成。此外,次要临床终点也得以实现,第12周时PASI 90的反应率为74.4%。PASI 75/90和PGA 0/1的反应在第52周得以维持,显示出显著且持久的疗效。
Stapokibart(凯美博济生物制药有限公司)
Stapokibart(康悦达®,Kangyueda,CM310)是一种人源化、铰链稳定的IgG4κ单克隆抗体,由凯美博济生物开发,靶向白细胞介素-4受体α(IL-4 Rα),阻断IL-4和IL-13信号传导,最终减少2型炎症。2024年9月12日,凯美博济生物宣布NMPA批准Stapokibart用于治疗中重度特应性皮炎。凯美博济还在寻求补充批准。2024年6月,Stapokibart用于治疗慢性鼻窦炎伴鼻息肉(CRSwNP)的新药上市申请被NMPA接受,并获得优先审评。2024年4月,NMPA接受了Stapokibart针对季节性过敏性鼻炎的新药上市申请。凯美博济还与石药集团合作,开发和商业化Stapokibart,用于治疗中重度哮喘、慢性阻塞性肺病及其他呼吸系统疾病。
NMPA的批准基于一项多中心、随机、双盲、安慰剂对照的III期验证性临床研究(CM310AD005)。该试验评估了Stapokibart在中重度特应性皮炎患者中的疗效、安全性、药代动力学/药效学(PK/PD)和免疫原性。共有500名合格患者按1:1比例随机分配到接受Stapokibart(600–300 mg Q2W)或安慰剂组。共同主要终点是治疗第16周时,从基线改善≥75%的湿疹面积和严重程度指数(EASI-75)的受试者比例,以及IGA评分为0或1且从基线降低≥2分的受试者比例。结果显示,经过16周的Stapokibart治疗后,达到EASI-75的受试者比例为66.9%,而达到IGA评分为0或1且降低≥2分的受试者比例为44.2%,均超过安慰剂组(分别为25.8%和16.1%),两组差异均有统计学意义(p < 0.0001)。两个共同主要终点均成功达成。在维持治疗期的第52周,Stapokibart组的EASI-75达成率为92.5%,而从安慰剂转为Stapokibart的受试者为88.7%;达到IGA评分为0或1且从基线降低≥2分的比例分别为67.3%和64.2%。
Ebronucimab(康方生物)
Ebronucimab(AK102,伊喜宁®)是一种人源抗PCSK9 IgG1λ单克隆抗体,由康方生物和东曜药业联合开发,用于治疗高胆固醇血症。2024年9月,NMPA批准Ebronucimab用于治疗原发性高胆固醇血症、混合型高脂血症和杂合子家族性高胆固醇血症(HeFH)。
该批准基于四项随机关键研究的结果,包括三项研究评估了Ebronucimab与安慰剂在治疗原发性高胆固醇血症和混合型高脂血症中的效果(CTR20212815 [450 mg Q4W剂量]、CTR20213363 [450 mg或600 mg Q6W剂量]、CTR20212466 [450 mg Q4W或150 mg Q2W剂量]),以及一项针对HeFH治疗的研究(CTR20191935 [450 mg Q4W、300 mg Q4W或150 mg Q2W剂量])。Ebronucimab在不同研究和不同适应症人群中显示出一致的疗效,能够显著降低低密度脂蛋白胆固醇(LDL-C)水平,每个剂量周期的最大降幅超过65%。在纳入原发性高胆固醇血症(包括HeFH)或混合型血脂异常患者的III期CTR20212466研究(NCT05255094)中,结果显示Ebronucimab在中国患者中具有显著的LDL-C降低效果,尤其是在接受450 mg Q4W或150 mg Q2W剂量的患者中。
Ongericimab(上海君实生物)
Ongericimab(JS002,昂戈瑞西单抗,君适达®)是由君实生物开发的一种人源化、铰链稳定的抗PCSK9 IgG4单克隆抗体。2024年10月11日,Ongericimab获得国家药品监督管理局(NMPA)批准,用于治疗原发性高胆固醇血症和混合型高脂血症(与他汀类药物联合使用)。此外,还有两项补充新药上市申请(sNDA)正在NMPA审查中,一项是用于治疗杂合子家族性高胆固醇血症,另一项是用于对他汀类药物不耐受或禁忌的原发性高胆固醇血症和混合型高脂血症患者(单药使用)。
此次批准主要基于两项关键性III期临床试验,均为多中心、随机、双盲、安慰剂对照试验:JS002–003(NCT04781114)和JS002–006(NCT05532800)。JS002–003研究评估了Ongericimab皮下注射治疗中国原发性高胆固醇血症和混合型高脂血症的疗效和安全性,共纳入806名受试者。结果显示,Ongericimab以150 mg每两周一次(Q2W)或300 mg每四周一次(Q4W)的剂量给药,可显著降低低密度脂蛋白胆固醇(LDL-C)水平,与安慰剂相比降幅超过60%,且这种降低在52周的治疗期内得以维持。此外,Ongericimab还显著改善了其他血脂指标,包括非高密度脂蛋白胆固醇、载脂蛋白B(ApoB)和总胆固醇。Ongericimab的安全性良好,在研究期间未出现新的安全性问题。
JS002–006研究旨在评估Ongericimab通过两种不同皮下注射装置(预充式注射器和自动注射器)给药治疗原发性高胆固醇血症和混合型高脂血症的疗效和安全性。该试验共纳入255名受试者。结果显示,两种给药方式均显示出显著的降脂效果。在接受150 mg Q2W剂量治疗12周后,预充式注射器组的LDL-C水平降低了72.7%,自动注射器组降低了71.1%,与安慰剂相比均显示出良好的安全性。
Enlonstobart(石药集团)
Enlonstobart(SG001,恩朗苏拜单抗,恩舒幸®)是一种人源化、铰链稳定的抗PD-1 IgG4单克隆抗体,通过阻断PD-1通路,使免疫细胞能够更有效地靶向并摧毁癌细胞。Enlonstobart最初由杭州森瑞博生物科技有限公司开发。2018年11月,森瑞博生物与石药集团达成战略合作,共同开发Enlonstobart。2024年6月,Enlonstobart获得NMPA附条件批准上市,用于治疗既往对铂类化疗无反应且PD-L1表达阳性(联合阳性评分[CPS]≥1)的复发或转移性宫颈癌患者。
Enlonstobart的批准主要基于一项关键性II期试验(NCT04886700)的数据,该试验显示在晚期宫颈癌治疗中客观缓解率(ORR)显著提高。在这项多中心、单臂、开放标签的研究中,共纳入107名宫颈癌患者,每名患者每两周接受一次240 mg Enlonstobart治疗,最大治疗持续时间为24个月,或直至疾病进展、无法耐受的毒性或其他研究终止标准出现。试验结果显示,ORR为29%,由独立影像学审查委员会评估,其中2名患者达到完全缓解,29名患者达到部分缓解。中位缓解持续时间(DoR)为16.6个月。此外,Enlonstobart显示出良好的安全性。
一项正在进行的III期临床试验(NCT05715840/CTR20230132)正在评估Enlonstobart联合铂类化疗(有或无贝伐珠单抗)作为复发或转移性宫颈癌患者的一线治疗方案,这些患者PD-L1表达阳性(CPS≥1)。此外,还有多项临床研究正在进行,以探索Enlonstobart联合纳米药物、抗体药物、抗体偶联药物(ADC)和小分子疗法在多种实体瘤治疗中的潜力。
Iparomlimab、Tuvonralimab(齐鲁普吉湾生物治疗公司)
Iparomlimab和Tuvonralimab(齐倍安®)分别是抗PD-1和抗CTLA-4单克隆抗体,是齐鲁普吉湾生物治疗公司开发的双抗体混合物PSB205(也称为QL1706)的成分。该混合物通过公司专有的MabPair技术由单个细胞系生产。Iparomlimab是一种铰链稳定的IgG4单克隆抗体,而Tuvonralimab是一种IgG1单克隆抗体,其重链和轻链均携带突变,以确保正确链的高效配对。
2024年9月,NMPA授予PSB205加速批准,用于治疗复发或转移性宫颈癌。此次批准基于齐鲁制药在中国开展的关键性单臂II期DUBHE-C-206研究(NCT05557565)的结果,该研究纳入了148名免疫检查点抑制剂初治的复发或转移性宫颈癌患者,这些患者既往对铂类化疗(有或无贝伐珠单抗)一线治疗失败。患者接受Iparomlimab和Tuvonralimab 5.0 mg/kg静脉注射,每三周一次(Q3W)。研究的主要终点是由独立审查委员会(IRC)评估的ORR,并持续评估长达2年。结果显示,ORR为33.8%,疾病控制率(DCR)为64.9%,中位无进展生存期(PFS)为5.4个月。一项正在进行的随机、双盲、安慰剂对照的III期试验(NCT05446883)正在评估Iparomlimab和Tuvonralimab联合化疗作为持续性、复发性或转移性宫颈癌患者的一线治疗方案,该试验的主要完成日期为2024年9月。
Iparomlimab和Tuvonralimab混合物还在研究用于治疗鼻咽癌(NPC)和非小细胞肺癌(NSCLC)。在NPC方面,一项单臂II期研究(NCT05576272)的结果显示,29名患者接受了QL1706(5 mg/kg静脉注射)联合化疗(吉西他滨和顺铂)治疗。主要终点是安全性和耐受性,次要终点包括ORR、PFS和总生存期(OS)。中位随访时间为15.5个月,中位ORR和PFS分别为82.1%和12.5个月,中位OS尚未达到。在NSCLC方面,一项开放标签、多队列II期研究(NCT05329025)的结果显示,91名晚期表皮生长因子受体(EGFR)野生型(60名患者)和EGFR突变型(31名患者)NSCLC患者接受了QL1706联合贝伐珠单抗、紫杉醇或培美曲塞以及卡铂治疗。所有药物均在每三周一次(Q3W)的第1天按协议规定剂量静脉注射,QL1706剂量为5 mg/kg。对于EGFR野生型NSCLC患者,ORR为45%,中位PFS为6.8个月。对于EGFR突变型NSCLC患者(既往接受过EGFR酪氨酸激酶抑制剂[TKI]治疗后进展),ORR为54.8%,中位PFS为8.5个月。
Benmelstobart(中国生物制药有限公司)
Benmelstobart(安度伟,TQB2450)是一种人源化抗PD-L1 IgG1κ单克隆抗体,其Fc区域经过工程改造(D265A),以降低效应功能。该药物由中国生物制药的子公司正大天晴药业开发。2024年4月29日,基于随机、双盲、安慰剂对照、多中心III期ETER701(NCT04234607)试验的结果,NMPA批准了Benmelstobart与安罗替尼、卡铂和依托泊苷联合用于广泛期小细胞肺癌(ES-SCLC)的一线治疗。
该研究比较了Benmelstobart联合安罗替尼和依托泊苷/卡铂(EC;n = 246)、安慰剂/安罗替尼联合EC(n = 245)或双安慰剂/EC单独治疗(n = 247)。Benmelstobart(1200 mg静脉注射)在每个21天周期的第1天给药。结果显示,Benmelstobart联合安罗替尼和EC治疗组的中位总生存期(OS)显著提高,达到19.3个月,而单独化疗(依托泊苷/卡铂)组为11.9个月(风险比HR 0.61,p < 0.05)。相比之下,安罗替尼联合EC组的OS改善并不显著(13.3个月对比11.9个月;HR 0.86;p = 0.1723)。
与单独EC组相比,Benmelstobart联合安罗替尼和EC组的中位无进展生存期(PFS)显著延长(6.9个月,95%置信区间6.2–8.3,对比4.2个月,95%置信区间4.17–4.24;HR 0.32,95%置信区间0.26–0.41;p < 0.0001),安罗替尼联合EC组的PFS也显著延长(5.6个月,95%置信区间5.6–6.8,对比4.2个月,95%置信区间4.17–4.24;HR 0.44,95%置信区间0.36–0.55;p < 0.0001)。
截至2024年7月,NMPA正在审查Benmelstobart用于另外两种适应症的上市申请:1)与安罗替尼联合用于治疗既往接受过一线或二线化疗方案但未能成功或不耐受的复发或转移性子宫内膜癌;2)与安罗替尼联合用于晚期不可切除或转移性肾细胞癌的一线治疗。Benmelstobart还在进行其他适应症的III期临床试验,包括非小细胞肺癌(NSCLC)放化疗后的维持治疗以及NSCLC的一线治疗。
Ivonescimab(康方生物、Summit Therapeutics)
Ivonescimab(依达方®)是一种四价双特异性抗体,靶向PD-1和人血管内皮生长因子(VEGF),经过工程改造以降低效应功能(L234A、L235A)。通过靶向这两种抗原,该药物将癌症免疫疗法与抗血管生成机制相结合。康方生物已将Ivonescimab在包括美国、加拿大、欧洲、日本、拉丁美洲、非洲和中东等地区的开发和商业化权利独家授权给Summit Therapeutics。迄今为止,Ivonescimab的晚期临床研究主要集中在非小细胞肺癌(NSCLC)适应症上。NMPA已授予Ivonescimab三项突破性疗法认定,用于不同场景和患者群体的NSCLC治疗。
2024年5月,NMPA批准了Ivonescimab联合化疗用于治疗表皮生长因子受体(EGFR)突变的局部晚期或转移性非鳞状NSCLC患者,这些患者在接受EGFR酪氨酸激酶抑制剂(TKI)治疗后病情进展。Ivonescimab是首个获得上市批准的靶向PD-1和VEGF组合的抗体。此次批准基于在中国进行的随机、双盲III期hARMONi-A研究(AK112–301,NCT05184712,CTR20213079)的结果。患者按1:1比例随机分配接受静脉注射Ivonescimab(20 mg/kg)联合培美曲塞(500 mg/m²)和卡铂(AUC 5)(n = 161)或静脉注射安慰剂联合化疗(n = 161),每三周一次,共四个周期。主要终点是由独立影像学审查委员会(IRC)根据实体瘤疗效评价标准(RECIST)v1.1在意向治疗(ITT)人群中评估的无进展生存期(PFS)。中位随访时间为7.89个月。Ivonescimab组的中位PFS为7.1个月(95%置信区间5.9–8.7),而安慰剂组为4.8个月(95%置信区间4.2–5.6),差异为2.3个月(风险比HR 0.46,95%置信区间0.34–0.62;p < 0.001)。Ivonescimab组的客观缓解率(ORR)为50.6%(95%置信区间42.6%–58.6%),而安慰剂组为35.4%(95%置信区间28.0%–43.3%),差异为15.6%(95%置信区间5.3%–26.0%;p = 0.006)。
2024年7月,康方生物向NMPA提交了Ivonescimab单药用于一线治疗PD-L1阳性(PD-L1 TPS ≥ 1%)局部晚期或转移性NSCLC的补充生物制品许可申请(sBLA)。该补充申请基于III期hARMONi-2(AK112–303,NCT05499390)研究的结果,该研究评估了Ivonescimab与抗PD-1药物帕博利珠单抗(Keytruda®)在一线治疗PD-L1表达阳性(PD-L1肿瘤比例评分[TPS] ≥ 1%)的晚期NSCLC患者中的疗效和安全性。患者(总n = 398)被随机分配接受Ivonescimab静脉注射或帕博利珠单抗200 mg每三周一次。研究的主要终点是由独立影像学审查委员会(IRRC)根据RECIST v1.1评估的PFS。2024年9月,hARMONi-2研究的预设中期分析数据的初步结果呈阳性。
在意向治疗(ITT)人群中,Ivonescimab组的中位PFS为11.14个月,而帕博利珠单抗组为5.82个月;Ivonescimab组的ORR为50.0%,而帕博利珠单抗组为38.5%。此外,在亚组分析中,Ivonescimab在多个因素(如年龄、性别、PD-L1表达、组织学类型以及肝或脑转移的存在)方面均优于帕博利珠单抗。
Sacituzumab Tirumotecan(四川科伦药业有限公司、默沙东公司)
Sacituzumab Tirumotecan(佳泰萊®,SKB264,MK-2870)是一种人源化IgG1κ抗体偶联药物(ADC),靶向TROP2,并携带一种专有的细胞毒性、贝洛替康衍生物载荷。这种ADC的平均药物抗体比(DAR)为7.4,包含一种水解连接子,能够在细胞外pH敏感性裂解和细胞内酶促裂解后释放膜可渗透的载荷。2022年,科伦药业将Sacituzumab Tirumotecan在中国大陆、香港、澳门和台湾以外地区的开发权授予默沙东公司。
Sacituzumab Tirumotecan已获得NMPA授予的三项突破性疗法认定,分别用于:1)局部晚期或转移性三阴性乳腺癌(TNBC);2)局部晚期或转移性HR阳性、HER2阴性乳腺癌,这些患者已接受过至少二线系统治疗;3)局部晚期或转移性EGFR突变非小细胞肺癌(NSCLC),这些患者在EGFR-TKI治疗后失败。
2024年11月21日,NMPA授予Sacituzumab Tirumotecan在中国的上市许可,用于治疗不可切除的局部晚期或转移性TNBC成人患者,这些患者已接受过至少两种先前的系统治疗,其中至少一种用于晚期或转移性环境。此次批准基于III期OptiTROP-Breast01研究的积极结果。
在III期OptiTROP-Breast01研究(NCT05347134)中,Sacituzumab Tirumotecan单药治疗显示出与化疗相比具有统计学显著性和临床意义的无进展生存期(PFS)和总生存期(OS)获益。该研究纳入了局部复发或转移性TNBC患者,这些患者之前接受过两种或更多治疗,包括至少一种用于转移性环境的治疗。患者被随机分配(1:1)到实验组(n = 130)和对照组(n = 133),按先前治疗线数(2–3对比>3)分层。然后比较了Sacituzumab Tirumotecan(5 mg/kg,每28天周期的第1天和第15天静脉注射)与对照组的效果,对照组为医生选择的化疗药物(艾日布林、长春瑞滨、卡培他滨或吉西他滨)。主要终点是由盲态独立中心评估(ICR)的无进展生存期(PFS)。TROP2表达通过免疫组化(IHC)使用半定量H评分法确定。基于中期分析(数据截止日期:2023年6月21日),PFS主要终点达到,进展或死亡风险降低69%(风险比HR 0.31;95%置信区间0.22至0.45;p < 0.00001)。由盲态ICR评估的中位PFS分别为5.7个月(95%置信区间4.3至7.2)和2.3个月(95%置信区间1.6至2.7),Sacituzumab Tirumotecan组和化疗组分别为此,6个月PFS率分别为43.4%和11.1%。在TROP2 H评分>200的患者亚组中,中位PFS分别为5.8个月和1.9个月(HR 0.28;95%置信区间0.17至0.48)。由盲态ICR评估的客观缓解率分别为43.8%和12.8%。
NMPA正在审查两项补充上市申请。2024年8月,NMPA接受了Sacituzumab Tirumotecan用于治疗局部晚期或转移性EGFR突变NSCLC患者的上市申请,这些患者在接受EGFR-TKI和铂类化疗后失败。另一项申请于2024年10月被NMPA接受,用于治疗局部晚期或转移性EGFR突变NSCLC成人患者,这些患者在接受EGFR-TKI治疗后病情进展。
Sacituzumab Tirumotecan用于治疗局部晚期或转移性EGFR突变NSCLC患者的上市申请基于OptiTROP-Lung03研究的结果。这项随机、关键性临床研究评估了Sacituzumab Tirumotecan单药治疗(5 mg/kg,每两周一次静脉注射)与多西他赛相比,用于治疗局部晚期或转移性EGFR突变NSCLC患者,这些患者在接受EGFR-TKI治疗和铂类化疗后失败。在预定分析中,Sacituzumab Tirumotecan单药治疗显示出与多西他赛相比在客观缓解率(ORR)和无进展生存期(PFS)方面的统计学显著性和临床意义的改善。
默沙东已启动了10项III期临床试验,评估Sacituzumab Tirumotecan的效果。其中五项研究正在招募NSCLC患者(NCT06074588、NCT06170788、NCT06312137、NCT06305754和NCT06422143),每项研究分别招募子宫内膜癌(NCT06132958)、HR+/HER2−不可切除的局部晚期或转移性乳腺癌(NCT06312176)、宫颈癌(NCT06459180)、三阴性乳腺癌(TNBC)(NCT06393374)和胃食管癌(NCT06356311)患者。
Seniprutug(BIOCAD)
Seniprutug(Tribuvia®,BCD-180)是一种人源化抗T细胞受体Vbeta9 IgG1单克隆抗体,由BIOCAD和俄罗斯国家研究医科大学(Pirogov)的科学家共同开发。2024年4月,俄罗斯联邦卫生部注册了Seniprutug(Tribuvia®),用于治疗放射学轴性脊柱关节炎(axSpA)患者。这是首个获得上市批准的靶向TCR Vbeta9的抗体。
在白俄罗斯和俄罗斯联邦进行的5臂III期LEVENTA研究(NCT06333210)中,比较了Seniprutug固定剂量与阿达木单抗和安慰剂在活动性axSpA患者中的效果。该研究纳入了HLA-B27+的放射学axSpA和非放射学axSpA患者,这些患者对非甾体抗炎药(NSAIDs)无反应,未接受过生物疾病修饰抗风湿药物(bDMARDs)或靶向合成疾病修饰抗风湿药物(tsDMARDs),以及那些对bDMARDs和/或tsDMARDs疗效不足和/或失去疗效的患者。研究组的干预措施为:1)静脉注射Seniprutug;2)静脉注射安慰剂;3)静脉注射Seniprutug+皮下注射安慰剂;4)静脉注射+皮下注射安慰剂;5)静脉注射安慰剂+皮下注射阿达木单抗。主要结果指标是在第24周时,bDMARDs和tsDMARDs初治患者以及bDMARDs和/或tsDMARDs经验患者中达到ASAS40的比例。ASAS40(脊柱关节炎评估40%)反应定义为在评估axSpA严重程度的评分系统中四个领域中的三个领域改善≥40%。
在II期ELEFTA研究(NCT05445076)中,Seniprutug显示出优于安慰剂的效果,该研究评估了两种剂量的Seniprutug与安慰剂在活动性放射学axSpA患者中的对比。该研究纳入了HLA-B27+的放射学axSpA患者,这些患者对NSAIDs无反应,未接受过生物治疗或tDMARDs。共260名患者被随机分为三组,分别接受5 mg/kg或7 mg/kg剂量的Seniprutug或安慰剂。Seniprutug在第0、12、36周给药。安慰剂组的患者在第24周转为5 mg/kg剂量的Seniprutug,并在第36周继续治疗。主要结果指标是在第24周时达到ASAS40的患者比例。接受7 mg/kg和5 mg/kg剂量Seniprutug的患者在第24周达到ASAS40的比例分别为51.4%和40.8%,而安慰剂组为24%(p = 0.0012和p = 0.0417)。Seniprutug治疗的耐受
表2. 正在接受各国监管审查的商业化赞助的单克隆抗体治疗药物的上市申请
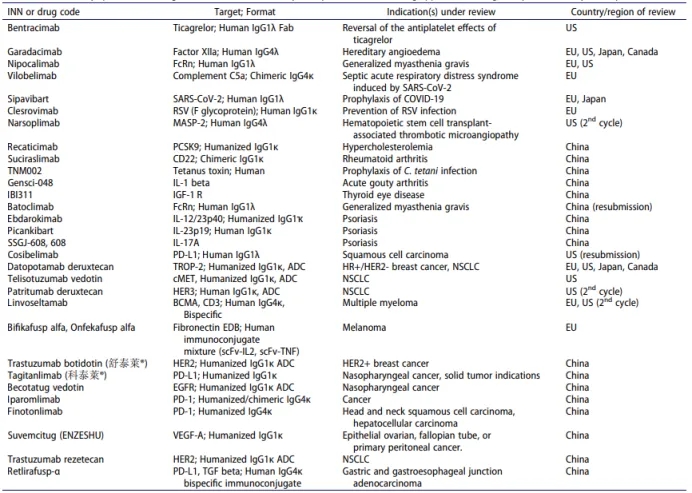
表中包含截至2024年12月9日在公共领域找到的信息。缩写:ADC,抗体药物偶联物;BCMA,B细胞成熟抗原;EDB,额外结构域B;HER,人表皮生长因子受体;IL,白细胞介素;MASP,甘露糖结合凝集素相关丝氨酸蛋白酶;NSCLC,非小细胞肺癌;PD-1,程序性细胞死亡蛋白1;scFv,单链可变片段;TNF,肿瘤坏死因子;TROP-2,滋养层细胞表面抗原2。欧洲药品管理局(EMA)正在评估的人用药品月度清单可在以下网址查看: https://www.ema.europa.eu/en/medicines/medicines-human-use-under-evaluation.。正在进行审查的抗体治疗药物的进一步数据可在以下网址查看https://www.antibodysociety.org/antibody-therapeutics-product-data/
Nipocalimab(强生公司)
Nipocalimab(JNJ-80202135,M281)是一种人源化IgG1λ单克隆抗体,靶向新生儿Fc受体(FcRn),该蛋白负责调节血液中IgG抗体的水平。通过阻断FcRn,Nipocalimab能够增加循环中IgG抗体的水平,从而减少自身免疫疾病中的免疫系统过度活跃。Nipocalimab正在开发用于治疗由致病性IgG抗体驱动的疾病,如全身性重症肌无力(gMG)和胎儿新生儿溶血病(HDFN)。
在一项双盲研究中,31.2%(24/77)接受Nipocalimab治疗的患者和13.2%(10/76)接受安慰剂的患者达到了预设的最小症状表达终点,即在任何时间点的MG-ADL总评分为0或1。正在进行的晚期临床研究也在评估Nipocalimab在以下患者中的疗效和安全性:1)患有gMG的儿童(II/III期NCT05265273研究);2)有HDFN风险的孕妇(III期AZALEA试验,NCT05912517);3)患有温抗体自身免疫性溶血性贫血的患者(II/III期ENERGY研究,NCT04119050);4)患有慢性炎症性脱髓鞘性多发性神经病的成人(II/III期NCT05327114研究);5)有胎儿和新生儿免疫性血小板减少症风险的孕妇(III期FREESIA1研究,NCT06449651)。截至2024年11月,这些研究正在招募受试者。
Vilobelimab(InflaRx N.V.)
Vilobelimab(Gohibic)是一种嵌合IgG4κ抗体,通过结合补体系统中的C5a成分来阻断补体介导的炎症反应。InflaRx N.V.正在开发Vilobelimab用于治疗由SARS-CoV-2引起的脓毒症急性呼吸窘迫综合征(ARDS)和溃疡性坏疽性脓皮病,这两种疾病中C5a信号传导起着关键作用。Vilobelimab获得了FDA的快速通道和孤儿药认定,以及EMA的孤儿药产品认定,用于治疗溃疡性坏疽性脓皮病。
2023年8月,EMA开始评估Vilobelimab的上市申请。2024年11月14日,EMA的人用药品委员会采纳了积极意见,建议批准Gohibic用于治疗正在接受全身性皮质类固醇治疗的SARS-CoV-2引起的ARDS成人患者。欧盟委员会的决定通常在意见采纳后67天内发布。
该申请包括了II/III期PANAMO试验(n = 368;NCT04333420)的结果,该试验评估了Vilobelimab联合标准治疗(SOC)与安慰剂联合SOC在治疗COVID-19相关严重肺炎中的效果。SOC包括联合使用皮质类固醇(97%)和抗凝剂(98%),以及免疫调节剂(约20%),主要是托珠单抗而非巴瑞替尼。对61名接受托珠单抗治疗的患者进行的事后亚组分析表明,Vilobelimab与托珠单抗联合使用可能在这些危重ARDS患者中产生协同生存益处。
Sipavibart(阿斯利康)
Sipavibart(AZD3152)是一种人源IgG1λ抗体,靶向SARS-CoV-2的刺突蛋白。该抗体源自SARS-CoV-2感染后患者的B细胞,经过优化以降低Fc效应功能(L234F、L235E、P331S),从而最小化抗体依赖性感染增强的风险,并延长半衰期(M252Y、S254T、T256E)。2022年5月,阿斯利康从RQ生物技术公司获得了Sipavibart的许可。阿斯利康向EMA提交了Sipavibart的上市申请,用于免疫受损患者的COVID-19暴露前预防。EMA对该申请的评估于2024年6月开始,该申请被接受为加速评估程序的一部分。阿斯利康还在日本提交了Sipavibart的上市申请,用于免疫受损患者的COVID-19暴露前预防。
该上市申请包括了I/III期SUPERNOVA试验(NCT05648110)的数据,该试验比较了Sipavibart与Tixagevimab/Cilgavimab(EVUSHELD)或安慰剂在预防免疫受损患者COVID-19中的效果。该研究包括一个开放标签的II期子研究,以评估AZD3152用于COVID-19暴露前预防的安全性、药代动力学(PK)和中和活性。在SUPERNOVA试验的第1天和第181天,AZD3152以300 mg剂量肌肉注射给药。共有1669/3335名参与者接受了Sipavibart,1666/3335名参与者接受了对照药物。试验结果显示,与对照组(Tixagevimab/Cilgavimab或安慰剂)相比,Sipavibart在免疫受损人群中显著降低了有症状COVID-19的发病率。该试验达到了两个主要终点:1)由任何SARS-CoV-2变异株引起的有症状COVID-19的相对风险降低;2)由不包含F456L突变的SARS-CoV-2变异株引起的感染的相对风险降低。
Clesrovimab(默沙东)
Clesrovimab(MK-1654)是一种人源单克隆IgG1κ抗体,由默沙东(MSD)开发,用于预防呼吸道合胞病毒(RSV)感染,特别是在婴儿和老年人等易感人群中。其作用机制是通过靶向病毒的F蛋白来中和RSV,该蛋白对于病毒进入宿主细胞至关重要。通过引入YTE突变(M252Y、S254T、T256E),其半衰期得以延长,从而可以每个RSV季节给药一次。EMA于2024年11月28日开始评估Clesrovimab的上市申请。
2024年10月,默沙东宣布了其IIb/III期临床试验(MK-1654-004,NCT04767373)的积极顶线结果,该试验比较了单剂量105 mg Clesrovimab肌肉注射与安慰剂在3632名健康早产儿和足月儿中的安全性和有效性。该研究的所有预设终点均得以实现。主要疗效终点是在给药后第150天(5个月)时,与安慰剂相比,RSV相关的需要≥1个下呼吸道感染或严重程度指标的医疗就诊的发病率降低了60.4%(95%置信区间:44.1至71.9,p < 0.001)。此外,在接受Clesrovimab治疗的参与者中,RSV相关住院(次要终点)和RSV相关下呼吸道感染住院(三级终点)在第150天时分别降低了84.2%(95%置信区间:66.6至92.6,p < 0.001)和90.9%(95%置信区间:76.2至96.5)。
默沙东正在III期MK-1654-007试验(NCT04938830)中评估Clesrovimab肌肉注射与Palivizumab肌肉注射在有严重RSV疾病风险的婴儿和儿童中的效果。该研究已招募约1000名受试者,截至2024年10月,该研究正在进行中,但不再招募受试者。该研究的主要完成日期为2025年4月。
Narsoplimab(Omeros公司)
Narsoplimab(OMS721)是一种人源抗甘露糖结合凝集素相关丝氨酸蛋白酶-2(MASP-2)抗体。该靶点是补体系统凝集素途径的效应酶。FDA授予了Narsoplimab突破性疗法认定,用于治疗高风险造血干细胞移植相关血栓性微血管病(TA-TMA)患者,并授予了孤儿药认定,用于预防补体介导的血栓性微血管病(TMA)和治疗TA-TMA。此外,Narsoplimab还被欧盟授予孤儿药产品认定,用于造血干细胞移植治疗。
Omeros公司为Narsoplimab提交的生物制品许可申请(BLA)滚动提交于2020年底完成。在收到FDA关于BLA的问题后,Omeros公司与FDA进行了持续讨论,内容包括:1)评估已有的临床试验数据、来自外部来源的历史对照人群数据、Narsoplimab扩展使用(即同情使用)计划的数据以及针对Narsoplimab作用机制的数据的分析计划;2)FDA对BLA重新提交的要求。公司已修订并重新提交了统计分析计划,FDA也已提出了对分析计划的进一步建议。在数据准备完成后,如果主要和次要疗效分析的结果支持重新提交,公司计划尽快完成并重新提交BLA。此外,公司计划在2025年上半年向EMA提交上市申请。
Omeros公司的临床研发管线还包括OMS1029,这是一种长效的下一代MASP-2抑制剂,目前正在I期研究中评估;以及Zaltenibart(OMS906),这是一种抗MASP-3抗体,正在PNH患者中进行II期研究。Omeros公司计划在2024年底前启动Zaltenibart在PNH中的III期项目。
Recaticimab(江苏恒瑞医药股份有限公司)
Recaticimab(SHR-1209,瑞卡西单抗)是一种长效、人源化IgG1单克隆抗体,选择性靶向PCSK9。其Fc区域经过突变(M252Y、S254T、T256E),以延长抗体在血清中的半衰期。2023年6月,恒瑞宣布其新药上市申请(NDA)被NMPA接受。该申请基于三项多中心、随机、双盲、安慰剂对照的III期临床试验(SHR-1209-301、SHR-1209-302和SHR-1209-303),分别针对不同的适应症。
SHR-1209-301研究(REMAIN-1,NCT04849000)
该研究评估了Recaticimab作为单药治疗非家族性高胆固醇血症和混合型高脂血症患者的疗效和安全性。主要终点是基线至第12周时低密度脂蛋白胆固醇(LDL-C)的变化百分比,对于150 mg每四周一次(Q4W)和450 mg每十二周一次(Q12W)的剂量方案,以及至第16周时300 mg每八周一次(Q8W)的剂量方案。共有703名患者随机分配接受Recaticimab(150 mg Q4W,n = 157;300 mg Q8W,n = 156;450 mg Q12W,n = 155)或安慰剂(n = 78、79和78)。与安慰剂相比,Recaticimab显著降低了患者的LDL-C水平,分别在150 mg Q4W、300 mg Q8W和450 mg Q12W剂量下实现了49.6%(95%置信区间:44.2%–54.9%)、52.8%(95%置信区间:48.3%–57.2%)和45.0%(95%置信区间:41.0%–49.0%)的降低(所有比较的p < 0.0001)。Recaticimab的安全性与安慰剂相当。在12周或16周的治疗后,Recaticimab组的患者继续治疗至第24周,而最初接受安慰剂的患者则转为接受相同剂量方案的Recaticimab。无论是24周的Recaticimab治疗方案,还是在安慰剂后的12周或8周的Recaticimab治疗,均显示出持续的疗效。
SHR-1209-302研究(REMAIN-2,NCT04885218)
该研究评估了Recaticimab作为附加疗法在非家族性高胆固醇血症(非FH)和混合型高脂血症患者中的疗效和安全性,共纳入689名受试者。患者随机接受Recaticimab(150 mg Q4W、300 mg Q8W和450 mg Q12W)或安慰剂,治疗周期为48周。主要疗效终点是从基线至第24周计算的LDL-C水平的变化百分比。结果显示,与安慰剂相比,Recaticimab显著且呈剂量依赖性地降低了LDL-C水平,相应剂量下的治疗差异分别为-62.2%、-59.7%和-53.4%。在第24周时,85.8%至94.5%接受Recaticimab治疗的患者达到了LDL-C目标水平,并且这些降低在48周的试验期内得以维持。
SHR-1209-303研究
该研究评估了Recaticimab与其他降脂疗法联合用于杂合子家族性高胆固醇血症患者的疗效和安全性。共有143名患者随机分配接受Recaticimab(n = 95)或安慰剂(n = 48)。在第12周时,Recaticimab组的平均LDL-C降低幅度为54.4%(95%置信区间:-57.9%至-50.8%),而安慰剂组为4.5%(95%置信区间:-9.4%至0.3%)。治疗差异为-49.8%(95%置信区间:-55.8%至-43.9%;p < 0.0001),显示出Recaticimab的显著影响。
Suciraslimab(SinoMab BioScience Limited)
Suciraslimab(SM03,舒西利单抗)是一种嵌合抗CD22 IgG1抗体,由SinoMab BioScience开发,用于治疗类风湿关节炎。Suciraslimab特异性结合CD22的构象表位,促进CD22从顺式配体结合转变为反式配体结合,反式配体结合的CD22可以抑制B细胞受体(BCR)引发的针对自身细胞的免疫反应。2023年9月,NMPA接受了Suciraslimab治疗类风湿关节炎的生物制品许可申请(BLA),截至2024年6月,该申请处于最终审查阶段。SinoMab计划进一步开发Suciraslimab,用于治疗系统性红斑狼疮(SLE)、阿尔茨海默病相关的轻度认知障碍(MCI)以及阿尔茨海默病。
TNM002(珠海Trinomab生物技术有限公司)
TNM002是一种人源单克隆抗体,靶向破伤风神经毒素(TeNT),为预防或治疗破伤风提供了新的治疗途径。通过Trinomab的专有HitmAb®技术平台开发,TNM002通过结合TeNT毒素,中和其对神经系统的毒性作用,设计用于肌肉注射。2023年12月6日,Trinomab宣布其TNM002的新药上市申请(NDA)被NMPA接受,并被授予紧急破伤风预防的优先审评资格。
一项多中心、随机、双盲、平行、阳性对照的III期临床试验(CTR20223067/NCT05664750)评估了TNM002与人破伤风免疫球蛋白(HTIG)肌肉注射预防破伤风的效果。主要终点是给药后与基线相比破伤风中和抗体效价的增加。TNM002实现了保护性水平的抗破伤风抗体,其效果和抗体持续时间均优于标准的250 IU HTIG。TNM002显示出良好的安全性、耐受性和低免疫原性,在I至III期试验中,其不良事件发生率与安慰剂或HTIG相当。
Gensci-048(长春金赛药业有限公司)
Gensci-048(genakumab,金纳单抗)是一种人源化单克隆抗体,靶向白细胞介素-1β(IL-1β)通路。IL-1β是一种关键的促炎细胞因子,参与多种炎症性疾病。通过特异性阻断IL-1β与其受体的结合,genakumab能够抑制炎症反应。2024年4月,GenSci宣布其genakumab的上市申请已被国家药品监督管理局(NMPA)接受,用于治疗急性痛风性关节炎。此外,genakumab还在进行其他适应症的临床研究,如全身性幼年特发性关节炎和间质性肺病。
GenSci开展了一项前瞻性、随机、多中心、活性对照的III期临床研究(GUARD-1,CTR20223136),涉及中国成年痛风发作患者,研究包括48周的治疗期和12周的安全性随访。患者按1:1比例随机接受单剂量genakumab 200 mg皮下注射(n = 157)或甲泼尼龙7 mg肌肉注射(n = 156),在新发作间隔超过2周时按需重新给药。研究结果显示,与甲泼尼龙相比,genakumab有效减轻疼痛并延缓新发作。主要终点包括基线时受累最重关节的疼痛强度变化,使用0–100 mm视觉模拟量表(VAS)在治疗后72小时评估,以及12周内首次新痛风发作的时间。genakumab在减少疼痛强度方面与甲泼尼龙相当,全分析集中疼痛评分平均差异为-3.32 mm(95%置信区间:-7.56至0.91),符合方案集中为-2.21 mm(95%置信区间:-6.49至2.07)。此外,genakumab显著延长了首次新痛风发作的中位时间,甲泼尼龙组为45天(genakumab数据不可估计)。
IBI311(信达生物制药有限公司)
IBI311是一种重组抗胰岛素样生长因子1受体(IGF-1R)抗体,由信达生物开发,用于治疗甲状腺眼病(TED)。通过结合在TED患者中过度表达的IGF-1R受体,IBI311阻断IGF-1R信号传导,减少炎症通路的激活。这一作用有助于减少与眼眶成纤维细胞(OFs)激活相关的透明质酸和其他糖胺聚糖的产生,缓解如突眼、复视和眼部充血等症状。2024年5月,NMPA接受了IBI311用于TED治疗的新药上市申请(NDA)。
该NDA的接受基于一项多中心、随机、双盲、安慰剂对照的II/III期临床研究RESTORE-1(CTR20223393)的积极结果。III期结果显示,主要终点成功达成,与安慰剂相比,接受IBI311治疗的患者在突眼、疾病活动度和生活质量方面有显著改善。在第24周时,IBI311组中研究眼从基线减少≥2 mm突眼且对侧眼未恶化≥2 mm的受试者比例显著高于安慰剂组。接受IBI311和安慰剂治疗的受试者应答率分别为85.8%和3.8%,显示出81.9%的显著差异(95%置信区间:69.8%至93.9%,p < 0.0001)。IBI311在关键次要终点方面也显示出与安慰剂相比的显著改善,包括总体应答率(研究眼从基线减少≥2 mm突眼且临床活动评分改善≥2的受试者百分比)、临床活动评分(CAS)为0或1的受试者百分比以及研究眼从基线的突眼平均变化。IBI311的整体安全性良好,未报告严重不良事件。
Batoclimab(Harbour BioMed(广州)有限公司、Roivant Sciences Ltd.、HanAll Biopharma)
Batoclimab(HBM9161,IMVT-1401,巴托利单抗)是一种人源IgG1λ抗FcRn单克隆抗体,其Fc区域经过工程改造,以降低效应功能(L234A,L235A)。该抗体最初由HanAll Biopharma开发,用于治疗IgG介导的自身免疫性疾病,包括全身性重症肌无力(gMG)和格雷夫斯病。HanAll Biopharma将Batoclimab授权给Harbour BioMed,后者随后与CSPC集团的全资子公司NBP Pharma达成协议,在大中华区共同开发Batoclimab。HanAll还将Batoclimab的开发权授权给Roivant Sciences,用于北美、拉丁美洲、瑞士和北非以及欧盟、英国和中东地区。Batoclimab获得了NMPA授予的突破性疗法认定,用于治疗成年重症肌无力患者。
2023年6月,Harbour BioMed向NMPA提交了Batoclimab用于治疗gMG的上市申请,随后在2024年6月补充了额外的安全性数据。该申请基于在中国进行的一项安慰剂对照的III期研究(NCT05039190)的积极结果,该研究显示每周皮下注射680 mg剂量的Batoclimab在gMG的主要和次要终点方面均显示出疗效。共有131名抗体检测呈阳性的患者接受了安慰剂或每周皮下注射680 mg剂量的Batoclimab治疗。每个治疗周期包括5周的治疗期,患者接受6剂安慰剂或药物,随后是4周的观察期(总共9周)。在接受安慰剂治疗的患者中,抗体阳性患者在第一个周期中Myasthenia Gravis Activities of Daily Living Scale(MG-ADL)持续改善率为31.3%(64名中的20名),而Batoclimab组为58.2%(67名中的39名)(比值比,3.45;95%置信区间,1.62–7.35;p = .001)。
Roivant Sciences的子公司Immunovant优先开发了IMVT-1402,这是一种下一代抗FcRn抗体,由于其独特的FcRn结合方式,可能会避免因Batoclimab引起的白蛋白减少和随后胆固醇水平升高,从而具有更有利的整体治疗特性。尽管如此,Immunovant目前正在对gMG患者(NCT05403541)、甲状腺眼病患者(NCT05524571、NCT05517421、NCT05517447)进行Batoclimab的III期研究,以及在慢性炎症性脱髓鞘性多发性神经病患者(NCT05581199)和格雷夫斯病患者(NCT05907668)中进行II期研究。
Ebdarokimab(Akeso Biopharma, Inc.)
Ebdarokimab(AK101,依若奇单抗)是由Akeso Biopharma开发的一种人源抗IL-12/23p40 IgG1单克隆抗体。通过靶向IL-12和IL-23共有的p40亚基,Ebdarokimab能够抑制这些细胞因子的信号传导,这些细胞因子参与炎症反应。Ebdarokimab正在开发用于治疗中重度斑块状银屑病。
2023年中期,Ebdarokimab用于治疗中重度斑块状银屑病的上市申请正在接受NMPA审查。Ebdarokimab的长期安全性和有效性在一项单臂III期研究(CTR20220206)中进行了评估,该研究涉及中国中重度斑块状银屑病患者。研究包括三个组:
在之前16周的双盲、安慰剂对照研究(CTR20212660)中接受Ebdarokimab治疗的患者在第16周继续接受Ebdarokimab 135 mg治疗,随后每12周进行维持治疗,直至第52周。之前研究安慰剂组的患者在本研究中第16/20周接受Ebdarokimab 135 mg治疗,随后每12周进行维持治疗,直至第52周。直接参与本研究的患者在第0/4周接受Ebdarokimab 135 mg治疗,随后每12周进行维持治疗,直至第52周。
在2024年阿姆斯特丹欧洲皮肤科和性病学大会上报告的结果显示,第1组在第16周时,银屑病面积和严重程度指数(PASI)改善≥75%(PASI75)和静态医生整体评估(sPGA)0/1的应答率分别为80.5%和66.0%。第1组患者的PASI75和sPGA0/1应答率在第52周时保持一致。第2组患者在第16周转换为Ebdarokimab后,PASI评分下降。第32周时,PASI75和sPGA0/1的应答率分别为81.4%和71.1%,并保持至第52周。第3组在第16周时,PASI75和sPGA0/1的应答率分别为69.5%和59.1%,与第1组结果一致。
Picankibart(信达生物制药有限公司)
Picankibart(IBI112)是由信达生物制药开发的一种抗IL-23p19 IgG1κ单克隆抗体,用于治疗银屑病或其他自身免疫性疾病患者。该分子通过中和IL-23的p19亚基,阻止IL-23与细胞表面受体结合及其随后的下游信号传导,从而阻断IL-23诱导的IL-17产生。Picankibart的Fc区域经过YTE突变(M252Y、S254T、T256E)改造,以延长半衰期。2024年9月,信达生物制药宣布其Picankibart用于治疗中重度斑块状银屑病的新药上市申请(NDA)已被NMPA接受。
该NDA的接受基于一项随机、双盲、安慰剂对照的III期CLEAR-1研究(NCT05645627)的积极结果,该研究评估了IBI112在不同剂量方案下治疗中重度斑块状银屑病患者的疗效和安全性。研究共纳入500名患者,按1:2:2的比例随机分配接受安慰剂或Picankibart 200 mg每四周一次(Q4W),在第0、4和8周给药,随后每12周给予200 mg或100 mg。研究的主要终点是第16周时从基线改善超过90%的银屑病面积和严重程度指数(PASI 90)的患者百分比,以及第16周时达到静态医生整体评估(sPGA)0(清晰)或1(几乎清晰)的患者百分比。两项主要终点均成功达成,接受Picankibart治疗的患者中,达到PASI 90和sPGA 0或1的比例显著高于接受安慰剂的患者(PASI 90:80.3%对比2.0%;sPGA 0/1:93.5%对比13.1%,均p < 0.0001)。Picankibart还显示出良好的安全性,并达到了所有关键次要终点,皮肤清洁水平在一年内保持较高水平。
SSGJ-608(上海阳光国健药业有限公司、三生国健药业股份有限公司)
SSGJ-608(608)是一种人源化单克隆抗体,靶向IL-17A,由三生国健和上海阳光国健药业开发,用于治疗自身免疫和炎症性疾病患者。2024年11月,三生国健向NMPA提交了SSGJ-608用于中重度斑块状银屑病的新药上市申请。
在中国中重度斑块状银屑病患者中进行的III期SSGJ-608-PsO-III-01研究(NCT05536726)已成功达到所有主要和次要终点。这项随机、双盲、安慰剂对照、平行组SSGJ-608-PsO-III-01研究评估了两种剂量方案的SSGJ-608与安慰剂在中国中重度斑块状银屑病患者中的效果。受试者接受以下治疗:
SSGJ-608 160 mg在第0周+80 mg每两周一次(Q2W,6个周期)+在维持期每四周一次(Q4W);SSGJ-608 160 mg每四周一次(Q4W,3个周期)+在维持期每八周一次(Q8W);在预定时间点接受安慰剂以保持盲态。
研究包括一个12周的诱导剂量期,主要终点在12周时测量,随后是一个随机、双盲、40周的维持剂量期。在维持剂量期,评估了治疗后的反应/缓解维持以及复发情况。
在第12周时,接受160 mg Q4W + 160 mg方案的患者中,银屑病面积和严重程度指数(PASI)75(表示受试者改善≥75%的比例)为95.1%,接受160 mg Q4W方案的患者为93.4%。12周的主要疗效数据显示出快速的应答率和疗效优势。在维持治疗期间,608的给药间隔延长至Q4W或Q8W,疗效保持在较高水平。SSGJ-608还在强直性脊柱炎和非放射学轴性脊柱关节炎患者中进行临床研究。
更多阅读:



长按屏幕识别二维码
打开手机扫描二维码