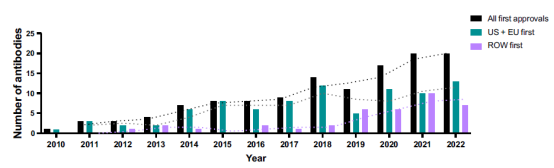
35
35
Donanemab(礼来公司)
Donanemab(Kisunla™,donanemab-azbt,LY3002813)是一种人源化IgG1k单克隆抗体,旨在靶向Aβ(p3-42),一种在聚集的淀粉样斑块中发现的淀粉样β的前体。FDA授予donanemab突破性疗法指定,用于治疗阿尔茨海默病。
2024年7月2日,美国食品药品监督管理局(FDA)批准Kisunla™(donanemab-azbt)用于治疗早期症状性阿尔茨海默病患者,包括轻度认知障碍(MCI)或轻度痴呆和确认的淀粉样病理。Kisunla的推荐剂量为每4周700毫克(Q4W)三次剂量,然后每4周1400毫克Q4W,通过静脉(IV)输注约30分钟给药。Kisunla的疗法由淀粉样斑块清除的证据支持。
2024年9月和10月,礼来公司宣布Kisunla在日本和英国分别获得批准。欧洲药品管理局(EMA)于2023年8月开始评估donanemab的市场申请;截至2024年11月,申请仍在评估中。
批准基于关键的第三阶段TRAILBLAZER-ALZ 2试验(NCT04437111)。该试验涉及1736名早期症状性阿尔茨海默病患者,这些患者有淀粉样β和低至中度tau病理。参与者被随机分配以1:1的比例接受IV donanemab(700毫克用于前三次剂量和1400毫克用于随后的剂量)或安慰剂(n = 860)Q4W 72周。该研究的主要结果是综合阿尔茨海默病评分量表(iADRS)从基线到76周的变化。结果显示,donanemab显著减缓了认知和功能下降,基于iADRS,某些亚组患者甚至获得了更大的益处。例如,在tau水平低至中等的患者中(n = 1182),donanemab治疗使iADRS下降了35%。
2024年10月29日,礼来公司宣布TRAILBLAZER-ALZ 6(NCT05738486)第三阶段研究的积极结果,显示使用水肿/积液(ARIA-E)在24周主要终点时,接受稍微修改剂量的donanemab治疗的参与者中,任何与治疗相关的成像异常有所减少。礼来公司正在与全球监管机构讨论研究结果,并计划为Kisunla提交潜在标签更新。礼来公司正在进行几项关于donanemab的临床试验,包括TRAILBLAZER-ALZ 3,该试验专注于减少有症状阿尔茨海默病风险的个体的淀粉样β病理,以及TRAILBLAZER-ALZ 5,这是一项针对中国、韩国、台湾和其他地区的早期症状性阿尔茨海默病患者的注册试验。
Axitinib(Syndax制药公司)
Axitinib(Nektivvo,axitinib-csf,SNDX-6352,UCB6352,INCIQ34176)是一种人源化、铰链稳定的(S228P)IgG4κ抗体,靶向集落刺激因子1受体(CSF-1R),该受体在单核细胞和巨噬细胞中表达。UCB于2016年授予Syndax axitinib的全球独家开发许可。2021年,Syndax和Incyte就axitinib在慢性移植物抗宿主病(cGVHD)和任何未来适应症的全球共同开发和商业化达成协议。Nektivvo将由Incyte和Syndax在美国共同商业化。Incyte在美国以外拥有Nektivvo的独家商业化权利。Axitinib被授予用于cGVHD治疗的孤儿药指定。
2024年8月14日,FDA批准axitinib-csf(Nektivvo)用于治疗至少40公斤体重的成人和儿童患者,这些患者在接受至少两种先前系统性治疗后cGVHD失败。推荐的Nektivvo剂量为0.3 mg/kg(更大35 mg)每2周(Q2W)在成人和体重40公斤及以上的儿童中,通过静脉(IV)输注超过30分钟给药。Axitinib是第一个获得市场批准的抗CSF-1R抗体。FDA的批准基于关键的第二阶段2 AGAVE-201研究(NCT04710576),该研究评估了241名接受至少两种先前系统性治疗的难治性cGVHD患者中三种剂量Nektivvo的安全性和有效性。患者被随机分配以1:1的比例接受IV donanemab(700 mg用于前三次剂量和1400 mg用于随后的剂量)或安慰剂(n = 860)Q4W 72周。该研究的主要结果是综合阿尔茨海默病评分量表(iADRS)从基线到76周的变化。结果显示,donanemab显著减缓了认知和功能下降,基于iADRS,某些亚组患者甚至获得了更大的益处。例如,在tau水平低至中等的患者中(n = 1182),donanemab治疗使iADRS下降了35%。
在治疗的前6个月中,donanemab显著减缓了认知和功能下降,中位反应时间为1.5个月。
Marstacimab (辉瑞公司)
Marstacimab(商品名:HYMPAVZI™,marstacimab-hnccp,PF-06741086)是一种人源化IgG1λ抗体,靶向组织因子途径抑制剂的Kunitz 2结构域。该靶标是一种天然存在的抗凝蛋白,可防止血栓形成。Marstacimab作为一种预防性药物,旨在预防或减少血友病患者的出血频率,已获得FDA的快速通道指定,用于预防或减少A型血友病伴抑制物或B型血友病患者(无论是否使用抑制物)的出血发作。EMA已授予marstacimab治疗血友病的孤儿药产品指定。Marstacimab于2024年在美国和欧盟获得批准。
2024年10月11日,FDA批准了HYMPAVZI™(marstacimab-hnccp)用于治疗A型或B型血友病的成人和青少年(无抑制物)。推荐的HYMPAVZI剂量包括300 mg(两次150 mg皮下注射),随后每周一次150 mg通过皮下注射(SC)维持剂量。批准基于3期BASIS试验(NCT03938792)的结果,该试验显示与常规预防和按需治疗相比,显著减少了出血。在研究中,HYMPAVZI在12个月的积极治疗期间,治疗出血的年化出血率分别比常规预防和按需治疗降低了35%和92%。HYMPAVZI的安全性与1/2期结果一致。HYMPAVZI是首个每周一次皮下注射的预防性治疗,适用于A型或B型血友病患者,也是首个通过预填充笔或注射器给药的预防性治疗。
欧盟委员会于2024年11月批准marstacimab用于12岁及以上、体重至少35公斤的严重血友病A(先天性VIII因子[FVIII]缺乏,FVIII < 1%)无FVIII抑制物或严重血友病B(先天性IX因子[FIX]缺乏,FIX < 1%)无FIX抑制物患者的常规预防出血发作。欧盟的营销授权基于关键的3期BASIS研究的结果。
Crovalimab(Chugai Pharmaceuticals、Genentech、F. Hoffmann-La Roche Ltd.)
Crovalimab(“派圣凯”,PiaSky,crovalimab-akkz,SKY59,RG6107,RO7112689)是一种补体C5抑制的、人源化IgG1k抗体,经过M428L/N434A修饰以增强在酸性pH下对新生儿Fc受体的亲和力,从而延长其血浆半衰期。由中外制药开发,crovalimab缺乏效应功能,并且作为治疗阵发性夜间血红蛋白尿(PNH)的药物。
Crovalimab利用中外制药的“循环抗体”技术开发,设计为能够反复结合其抗原,允许在低皮下剂量(SC)下每四周(Q4W)持续补体抑制。与现有的抗体药物不同,crovalimab靶向C5上的不同表位,为具有特定C5基因突变的PNH患者提供了潜在的替代选择。
2024年2月6日,SC crovalimab在中国首次获批用于治疗未接受过补体抑制剂治疗的PNH患者(年龄≥12岁)。这一批准基于第三阶段研究COMMODORE 2(NCT04434092)和COMMODORE 3(NCT04654468)在PNH患者中的积极结果。Crovalimab随后于2024年3月在日本获批,用于治疗PNH患者,基于第三阶段研究COMMODORE 1(NCT04432584)和COMMODORE 2在2024年6月和8月在美国和欧盟的积极结果。
全球第三阶段COMMODORE 1和2研究评估了crovalimab与依库珠单抗在PNH患者中的疗效和安全性。COMMODORE 1专注于C5抑制剂经验丰富的患者,而COMMODORE 2涉及未接受过补体抑制剂治疗的患者。COMMODORE 1评估了crovalimab的安全性、药代动力学(PK)、药效学(PD)和探索性疗效。这项研究的数据支持crovalimab的有利效益-风险概况,强调了SC给药的便利性和自我管理的选择。
COMMODORE 2研究的数据显示,与Q2W静脉注射依库珠单抗相比,SC crovalimab在控制疾病方面不劣,且对未接受过C5抑制剂治疗的患者具有可比的安全性。
COMMODORE 3在中国进行,是一项多中心、单臂试验,评估了C5抑制剂初治PNH患者(n=51)中的crovalimab。患者接受了基于体重的剂量方案,初始负荷阶段(IV)在第1天和第4天,每周皮下注射(SC)从第2天开始,第从5周开始进行维持SC Q4W。对于有临床益处的患者,治疗延长至24周以上。达到的主要终点是溶血控制和输血避免(TA),分别为78.7%在第5至25周实现溶血控制,TA率从治疗前的0.0%增加到治疗后的51.0%(p < 0.0001)。
研究中采用了10 mg每两周一次(Q2W)的剂量方案。结果显示,在这一剂量下,IMDELLTRA(n = 99)显示出显著的客观缓解率(ORR)为40%(95%置信区间:31%,51%),中位缓解持续时间(DoR)为9.7个月(置信区间:2.7个月,20.7+个月)。中位总生存期(OS)为14.3个月,最终生存数据仍在等待中。该治疗的安全性可管理,细胞因子释放综合征(CRS)是最常见的不良事件。2024年9月,安进公司(Amgen)公布了DeLLphi-301 II期研究的扩展随访数据,显示IMDELLTRA在之前接受过铂类化疗的广泛期小细胞肺癌(ES-SCLC)患者中具有持续的抗癌活性和可管理的安全性。
Tarlatamab正在多项研究中进行评估,包括DeLLphi-303(NCT05361395),一项Ib期研究,评估tarlatamab与标准治疗(SOC)联合用于一线ES-SCLC;DeLLphi-304(NCT05740566),一项随机III期试验,比较tarlatamab单药治疗与SOC化疗在小细胞肺癌(SCLC)二线治疗中的效果;DeLLphi-305(NCT06211036),一项随机III期试验,比较tarlatamab联合度伐利尤单抗与单独使用度伐利尤单抗作为ES-SCLC一线维持治疗的效果;DeLLphi-306(NCT06117774),一项随机、安慰剂对照的III期试验,评估tarlatamab在局限期小细胞肺癌同步放化疗后的效果;以及DeLLpro-300,一项Ib期研究,评估tarlatamab在原发性或治疗相关神经内分泌前列腺癌中的应用。
Zanidatamab(百济神州、Jazz Pharmaceuticals Plc、Zymeworks Inc)
Zanidatamab(ZW25)是一种人源化、双特异性IgGκ抗体,由半抗体(Fab-h-CH2-CH3 × scFv-h-CH2-CH3)组成,能够靶向HER2的两个非重叠表位。2022年,Zymeworks将Zanidatamab在美国、欧洲、日本以及除之前已授权的亚太地区以外的所有地区的开发和商业化权利授权给Jazz Pharmaceuticals。2018年,百济神州获得了Zanidatamab在亚洲(除日本外)、澳大利亚和新西兰的独家开发和商业化权利。美国食品药品监督管理局(FDA)授予了Zanidatamab突破性疗法认定,用于既往接受过治疗的HER2基因扩增胆道癌(BTC)患者;授予了Zanidatamab单药治疗难治性BTC的快速通道认定;以及授予了Zanidatamab在难治性BTC中的孤儿药认定。
2024年11月20日,Jazz Pharmaceuticals宣布FDA授予Ziihera®(zanidatamab-hrii)50 mg/mL注射液加速批准,用于治疗既往接受过治疗的不可切除或转移性HER2阳性(IHC 3+)胆道癌(BTC)成人患者,该适应症的诊断需通过FDA批准的检测手段进行确认。
一项验证性、随机III期试验(HERIZON-BTC-302;NCT06282575)正在评估Zanidatamab联合标准治疗(SOC)与SOC单独治疗在HER2阳性BTC患者一线治疗中的效果,该试验于2024年7月启动,目前正在招募患者。此次批准部分基于一项单臂、IIb期试验(HERIZON-BTC-01;NCT04466891)的结果,该试验在既往接受过吉西他滨治疗且病情进展的HER2扩增、不可切除、局部晚期或转移性BTC患者中评估了Zanidatamab的效果。患者(n = 87)接受了Zanidatamab 20 mg/kg静脉注射,每两周一次(Q2W)。患者根据HER2免疫组化(IHC)评分分为两组:队列1(IHC 2+或3+;HER2阳性)和队列2(IHC 0或1+)。主要终点是通过独立中心评估(ICR)在队列1中确认的客观缓解率。中位随访时间为12.4个月(四分位间距9.4–17.2)。在队列1中,33名患者(41.3%;95%置信区间30.4%–52.8%)确认了客观缓解,而队列2中没有HER2低表达患者出现缓解。
Zanidatamab作为胆道癌(BTC)治疗的上市申请也已提交至欧盟和中国。百济神州预计中国国家药品监督管理局(NMPA)将在2025年下半年批准Zanidatamab作为HER2+ BTC的二线治疗药物。
除了胆道癌,Zanidatamab还在进行III期研究,用于治疗HER2+乳腺癌(NCT06435429)和胃癌(NCT05152147)。
Zenocutuzumab(MCLA-128)是一种人源化、增强抗体依赖性细胞毒性(ADCC)的双特异性IgG1κ抗体,能够同时靶向生长因子受体HER2和HER3,从而阻断由HER3与其配体神经调节蛋白1(NRG1)或NRG1融合蛋白结合引起的信号传导。该抗体由Merus开发,用于治疗携带NRG1基因融合(NRG1+癌症)的实体瘤患者,这些患者通常对传统全身治疗反应不佳。美国食品药品监督管理局(FDA)授予了Zenocutuzumab突破性疗法认定,用于治疗既往接受过全身治疗后病情进展或无满意替代治疗选择的晚期不可切除或转移性NRG1+胰腺癌患者;授予了Zenocutuzumab快速通道认定,用于治疗接受标准治疗后病情进展的转移性NRG1+癌症患者;以及授予了Zenocutuzumab孤儿药认定,用于胰腺癌治疗。
2024年12月4日,Merus N.V.宣布FDA批准了BIZENGRI®(zenocutuzumab-zbco)用于治疗成人胰腺腺癌或非小细胞肺癌(NSCLC),这些患者为晚期不可切除或转移性且携带NRG1基因融合,并且在既往全身治疗后病情进展。此次批准基于开放标签的I/II期eNRGy试验(NCT02912949)的数据,该试验评估了Zenocutuzumab单药治疗在NRG1+ NSCLC、胰腺导管腺癌(PDAC)和其他实体瘤中的安全性和抗肿瘤活性。研究包括三个患者队列:1)NRG1+ NSCLC;2)NRG1+ PDAC;3)其他NRG1+实体瘤。在eNRGy研究的剂量扩展期间,参与者接受了每两周一次(Q2W)的750 mg Zenocutuzumab静脉输注。通过盲态独立中心评估(ICR)根据RECIST v1.1标准测量反应率。NRG1+ NSCLC队列(n = 64)和NRG1+ PDAC队列(n = 30)的客观缓解率(ORR)分别为33%(95%置信区间,22%–46%)和40%(95%置信区间,23%–59%),NRG1+ NSCLC队列的中位缓解持续时间(DOR)为7.4个月(95%置信区间,4.0–16.6),NRG1+ PDAC队列的DOR范围为3.7个月至16.6个月。
Odronextamab(Ordspono™,REGN1979)是一种人源化、铰链稳定的抗CD20×CD3双特异性IgG4κ抗体。其Fc区域经过工程改造(E233P、F234V、L235A、G236del),以降低效应功能。Regeneron利用其VelocImmune®技术和Veloci-Bi®平台开发了这种抗体,目前公司正在将其开发用于治疗复发/难治性(RR)B细胞非霍奇金淋巴瘤(NHL),包括滤泡性淋巴瘤(FL)和弥漫大B细胞淋巴瘤(DLBCL)。Odronextamab获得了美国食品药品监督管理局(FDA)的快速通道认定,以及FDA和欧洲药品管理局(EMA)分别授予的孤儿药和孤儿药产品认定,用于治疗滤泡性淋巴瘤(FL)和弥漫大B细胞淋巴瘤(DLBCL)患者。Regeneron在美国和欧盟提交了Odronextamab用于FL和DLBCL的上市申请。
基于EMA的积极意见,欧盟委员会于2024年8月批准了Odronextamab,用于治疗接受过两线或以上系统治疗后复发/难治性(R/R)的滤泡性淋巴瘤(FL)或弥漫大B细胞淋巴瘤(DLBCL)成人患者。这一批准得到了I期ELM-1(NCT02290951)和关键性II期ELM-2(NCT03888105)试验结果的支持,这些试验显示Odronextamab在R/R FL或R/R DLBCL成人患者中具有强大且持久的反应率,并且安全性可接受。在ELM-2研究中入组的R/R FL患者(n = 128)中,客观缓解率(ORR)为80%;其中73%的患者达到完全缓解(CR),中位缓解持续时间(DoR)为25个月。在嵌合抗原受体T细胞(CAR-T)治疗初治的R/R DLBCL患者中,入组ELM-2研究(n = 127)的ORR为52%,其中31%达到CR,完全缓解者的中位DoR为18个月。
2024年3月,FDA针对Odronextamab在R/R FL和R/R DLBCL(均接受过两线或以上系统治疗)的生物制品许可申请(BLA)发出了完整回复函(CRL)。唯一的问题涉及验证性试验的入组状态。Regeneron正在赞助多项作为OLYMPIA项目一部分的Odronextamab III期试验,这些试验涉及淋巴瘤患者。FDA要求这些试验包括剂量探索和验证性两部分。尽管剂量探索部分的入组已经开始,但CRL指出,这些试验的验证性部分应该正在进行中,并且在重新提交之前需要商定完成的时间表。截至2024年11月初,Regeneron尚未公开披露有关这些时间表的更新信息。



长按屏幕识别二维码
打开手机扫描二维码